








Serviços Personalizados
Journal

Artigo
 texto em
texto em  Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Pan-Amazônica de Saúde
versão impressa ISSN 2176-6215versão On-line ISSN 2176-6223
Rev Pan-Amaz Saude v.1 n.3 Ananindeua set. 2010
http://dx.doi.org/10.5123/S2176-62232010000300017
Informe de caso: enfermedad de Niemann-Pick con manifestaciones de insuficiencia hepática
Ivanete do Socorro Abraçado Amaral; Lizomar de Jesus Maués Pereira Moia; Erica Furtado Azevedo Coelho; Zilene Lameira de Medeiros; Maria de Fátima Pombo Montoril; Marialva Tereza Ferreira Araujo
Fundação Santa Casa de Misericórdia do Pará, Belém, Pará, Brasil
Endereço para correspondência
Correspondence
Dirección para correspondencia
Título original: Relatório de caso: doença de Niemann-Pick com manifestações de insuficiência hepática. Traducido por: Lota Moncada
RESUMEN
OBJETIVO: Relatar un caso de paciente
portadora de la enfermedad de Niemann-Pick que evolucionó con insuficiencia
hepática crónica, y, de ese modo, llamar la atención para
esta entidad nosológica.
RELATO DE CASO: Paciente mujer, 34
años de edad, presentando esplenomegalia, encaminada al hospital de
Fundação Santa Casa de Misericórdia do Pará, en
la Ciudad de Belém, Estado de Pará, Brasil, con diagnóstico
de enfermedad de Niemann-Pick, confirmada por dosaje
enzimático para investigación de enfermedad hepática.
La paciente presentaba cirrosis que evolucionó cinco años
después hacia insuficiencia hepática crónica descompensada,
habiendo sido encaminada para transplante de hígado.
CONSIDERACIONES FINALES: Se destaca que esta enfermedad, a pesar de
rara, puede ser una de las causas de la
enfermedad hepática crónica, y debe ser diagnosticada precozmente,
visto que la expectativa de vida del paciente
depende de la importancia y repercusión de los síntomas.
Palabras clave: Enfermedades de Niemann-Pick; Esfingomielina Fosfodiesterasa; Esplenomegalia.
INTRODUCCIÓN
La enfermedad de Niemann-Pick (ENP) es una enfermedad rara, de almacenamiento lisosómico o acumulación, en que la deficiencia de una enzima específica resulta en la acumulación de esfingomielina, un producto del metabolismo de las grasas, sobre todo con relación a fibroblastos. Se considera una enfermedad generalizada y grave. Esta enfermedad presenta actualmente seis subtipos, dependiendo de la gravedad de la deficiencia enzimática: subtipo A, la forma neuropática aguda; subtipo B, la forma visceral; subtipo C, la forma neuropática crónica; subtipo D, la variante nueva escocia; subtipo E, la forma adulta; y subtipo F, la enfermedad del histiocito azul marino1,2. En su forma juvenil grave, la enzima está completamente ausente, lo que conduce al desarrollo de alteraciones serias del sistema nervioso, porque los nervios no consiguen utilizar la esfingomielina para producir la mielina necesaria para las vainas que, normalmente, envuelven muchos nervios3. Un estudio observó que la distribución geográfica del subtipo B tiene mayor incidencia en Francia (Europa), Estados Unidos y Canadá (América del Norte), Brasil (América del Sur) y Japón (Asia)4.
La ENP se diagnostica por medio de la anamnesis y un examen físico detallado, apoyados por la biopsia de médula ósea con hallazgo de células de Niemann-Pick característicos, los histiocitos azul marino. En los Estados Unidos, el subtipo A ocurre con mayor frecuencia, abarcando cerca de 85% de los casos de esta enfermedad; y se estima que el subtipo C afecte a cerca de 500 niños. La ENP abarca a recién nacidos, niños y adultos de ambos sexos. El subtipo A se manifiesta en los primeros meses de vida, el paciente muere en la infancia; en el subtipo B, los primeros síntomas aparecen en la infancia, de forma no neuropática, sin embargo, muchos sobreviven hasta la edad adulta; y los subtipos C y D presentan disturbios neurodegenerativos, con inicio en los primeros 2 años o más de vida, o, aún, al final de la infancia5,6.
OBJETIVO
Relatar un caso clínico de paciente con la ENP que evolucionó para insuficiencia hepática crónica descompensada.
RELATO DE CASO
Mujer soltera, con actividad restricta al hogar, nivel de enseñanza primaria incompleta, natural del Municipio de Acará, procedente de Santa Izabel de Pará, Brasil, contaba 26 años cuando fue atendida por primera vez en el ambulatorio de hígado del hospital de la Fundação Santa Casa de Misericórdia do Pará (FSCMP), en la Ciudad de Belém, Estado de Pará, Brasil, en setiembre del 2002, encaminada por la Fundação Centro de Hemoterapia e Hematologia do Pará (HEMOPA) con diagnóstico de ENP Presentaba una dosis de esfingomielina, de leucocitos y fibroblastos, disminuida: 0,12 nanomoles/h/mg/proteína (0,74 a 4,91) y 1,9 nanomoles/h/mg/proteína (42 a 72), respectivamente. En el HEMOPA se realizaron los dosajes de P-glucosidasa y quitotriosidasa, los que fueron normales, y el mielograma, cuyo resultado mostró presencia de gran número de macrófagos intersticiales en la serie mono plasmocitaria, compatible con síndrome del histiocito azul marino o ENP.
En este período inicial, llamaban la atención la esplenomegalia (bazo tipo IV) y los marcadores serológicos para los virus de las hepatitis B y C, que eran negativos. En la primera consulta, presentaba turgescencia yugular, hepatoesplenomegalia, máculas hipocrómicas e hipercrómicas, de límites imprecisos, en los miembros inferiores. En la anamnesis de antecedentes mórbidos personales (AMP), la madre de la paciente refirió que, desde los tres años de vida, la paciente presentaba un abdomen aumentado y, en la de los antecedentes mórbidos familiares (AMF), la paciente informó que tiene cinco hermanos saludables. Con relación a los hábitos, negó uso de alcohol, tabaquismo o uso de drogas ilícitas.
En la primera consulta, el año 2002, al examen físico, la paciente se mostró consciente, orientada en el tiempo y el espacio, con presión arterial 140x80 mmHg, frecuencia cardíaca de 90 pulsaciones por minuto, ictericia (++/+4), manchas hipercrómicas e hipocrómicas en los miembros inferiores, adelgazada, con altura de 1,49 m, peso de 45 kg, bazo tipo IV e hígado a 2 cm del reborde costal derecho. Se realizó un examen neurológico con resultado normal. En este período, cuando sometida a biopsia de hígado en la FSCMP, presentó tiempo de actividad de la protrombina (TAP) a 100%; y el estudio anatomopatológico, realizado por el Departamento de Patología de la Universidade Federal do Pará, evidenció ENP por la presencia de células esponjosas, además de cirrosis. La endoscopia digestiva alta (EDA) no reveló várices esofágicas. Los exámenes de laboratorio resultaron en: hematíes: 3,9 millones/mm3, Hb 9,8g%, conteo de plaquetas 160.000/mm3, leucocitos 6.100/mm3, urea 33 mg/dL, creatinina 0,9 mg/dL, GGT 65 U/mL, glucosa 95 mg/dL, proteínas totales 7,6, albúmina 4,0, globulina 3,6, AST 92 U/mL, ALT 67 U/mL, alfa fetoproteína 2 ng/mL. En la densitometría ósea, demostró osteoporosis de columna lumbar y osteopenia de cuello de fémur, habiéndosele prescrito alendronato de sodio, 70 mg semanal, calcio y vitamina D diariamente.
La paciente dejó de comparecer al ambulatorio y, al volver, el año 2005, presentaba bazo a nivel de la cresta ilíaca izquierda, hematíes 4,6 millones/mm3 Hb 11,5 g/dL, Htc 36%, plaquetas 113.000/mm3, leucocitos 4.600/mm3, TAP 75%, TP 14", INR 1.1, creatinina 0,5, hierro sérico 60, FA 76, GGT 55, glucosa 90, TGO 61, TGP 25. En el EDA la paciente presentó un cordón varicoso de pequeño calibre, siendo prescrito propranolol.
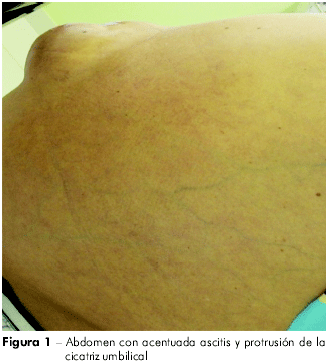
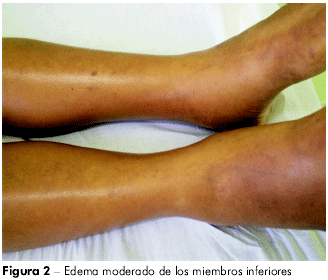
La paciente se ausentó por un tiempo más, retornando con descompensación del cuadro, con ascitis (Figura 1) y adelgazamiento, habiendo sido internada varias veces. En mayo del 2009, cuando se internó en el hospital de la FSCMP, presentaba ascitis de gran volumen, circulación colateral, edema de miembros inferiores (Figura 2) e incomodidad respiratoria; usaba espironolactona, propranolol, lactulosa y vitamina K. En los exámenes, presentó: leucocitos 3,2, Hb 11,8, glucosa 97, plaquetas 77.000, BT 1.94, BD 0,5, BI 1.42, FA 75, GGT 100, creatinina 0,70, urea 29, proteínas totales 6,2, albúmina 2,6, globulina 3,6, Na 140, K 3,5, AST 14, ALT 50, TAP 24", AP 30,2%, INR 2,07, MELD = 1 7. Se la sometió a tomografía de abdomen, en junio del 2009, la que indicó ascitis voluminosa, hígado poco aumentado a expensas de lóbulo hepático izquierdo de contornos lobulados, presentando parénquima con densidad finamente heterogénea; bazo de volumen acentuadamente aumentado, con densidad homogénea en la tomografía e imagen sugestiva de circulación colateral entre el bazo y el riñón. Una tomografía de tórax, realizada en junio del 2009, mostró pulmones aumentados de volumen, señalando áreas con diferencia de atenuación.
La paciente recibió alta hospitalaria con mejoría del cuadro clínico y fue encaminada para acompañamiento clínico en el ambulatorio de hígado de la FSCMP, y, de este, a programación para transplante de hígado. No se observó implicación neurológica, siendo sugerida ENP subtipo B.
DISCUSIÓN
La ENP subtipo B en el adulto es rara, afecta igualmente a ambos sexos, puede ocurrir en todas las poblaciones con incidencia de 1 para 250 mil personas. Los individuos afectados por este subtipo, en su mayoría, alcanzan la edad adulta sin comprometimiento neurológico o de la capacidad intelectual5,7. Estas descripciones evidencian lo raro del caso relatado.
El subtipo B es una enfermedad autosómica recesiva, resultante de la mutación del gen SPDM1, presente en el cromosoma 11 (p15,1 a p15,4), cromosoma 11p15, 1 a p15, 48, responsable por la deficiencia da esfingomielina ácida, pero con acumulación de esfingomielina apenas en el sistema de los monocitos macrófagos, y, debido al fenotipo (subtipo B), ocurren síntomas viscerales5,1. En el presente caso, los exámenes de ensayo enzimático individualizaron la reducción de la enzima en cuestión, con síntomas viscerales evidentes en la paciente. Entre las manifestaciones más frecuentes de la ENP subtipo B, se encontraron, baja estatura, hiperesplenismo, pancitopenia y alteraciones de la función hepática6,9.
La presencia de ascitis de moderada intensidad, circulación colateral, disturbio de coagulación y la presencia de cordón varicoso en el esófago son señales de cirrosis hepática y de gravedad en la EPN del subtipo B5. La esplenomegalia y/o hepatomegalia, en ese subtipo, suele ser detectada en los primeros años de vida, durante examen físico de rutina; sin embargo, en los casos muy leves, el aumento es sutil, y, por consiguiente, sólo es perceptible en la adolescencia o en la vida adulta6. La paciente en cuestión tuvo un diagnóstico tardío, a los 25 años de edad, aunque su madre haya referido que desde la infancia ya había observado el aumento de volumen del abdomen.
CONSIDERACIONES FINALES
O caso relatado se enquadrou na literatura como a ENP subtipo B no que tange aos achados clínicos, laboratoriais e histopatológicos. Ressaltamos a importância dessa enfermidade, a qual pode ser diagnosticada ainda na infância e pode ter evolução desfavorável, levando o paciente a transplante de fígado.
REFERENCIAS
1 Pavlu-Pereira H, Asfaw B, Poupetová H, Ledvinová J, Sikora J, Vanier MT, et al. Acid sphingomyelinase deficiency: phenotype variability with prevalence of intermediate phenotype in a series of twenty-five Czech and Slovak patients. A multi-approach study. J Inherit Metab Dis. 2005;28(2):203-27. DOI:10.1007/s10545-005-5671-5 [ Links ]
2 Garver WS, Francis GA, Jelinek D, Shepherd G, Flynn J, Castro G, et al. The National Niemann-Pick C1 disease database: report of clinical features and health problems. Am J Med Genet A. 2007 Jun;143A(11):1204-11. DOI:10.1002/ajmg.a.31735 [ Links ]
3 Spence M, Callahan J. Sphingomyelin-cholesterol lipidoses: the Niemann-Pick group of diseases. New York: McGraw-Hill; 1989.
4 Calogera MS, Desnick RJ, McGovern MM, Wasserstein MP, Schuchman EH. The demographics and distribution of type B Niemann-Pick disease: novel mutations lead to new genotype/phenotype correlations. Am J Hum Genet. 2002 Dec;71(6):1413-9. [ Links ]
5 Schwartz RAl. Niemann-Pick disease. New Jersey: Medical School; 2008.
6 Goldman L, Ausiello D. Tratado de medicina interna. 21. ed. Rio de Janeiro: Guanabara Koogan; 2001. 1231 p.
7 Schuchman EH, Desnick RJ. Niemann-Pick disease types A and B: acid sphingomyelinase deficiencies. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill; 2001. p. 3589-610.
8 Takahashi T, Suchi M, Desnick RJ, Takada G, Schuchman EH. Identification and expression of five mutations in the human acid sphingomyelinase gene causing types A and B Niemann-Pick disease: molecular evidence for genetic heterogeneity in the neuronopathic and non-neuronopathic forms. J Biol Chem. 1992 Jun;267(18):12552-8. [ Links ]
9 Wasserstein MP, Desnick RJ, Schuchman EH, Hossain S, Wallenstein S, Lamm C, et al. The natural history of type B Niemann-Pick disease: results from a 10-year longitudinal study. Pediatrics. 2004 Dec;114(6):672-7. DOI:10.1542/peds.2004-0887 [ Links ]
 Correspondência
/ Correspondence/ Correspondencia:
Correspondência
/ Correspondence/ Correspondencia:
Erica Furtado Azevedo Coelho
Conjunto Mendara
I, rua J, no 110
Tel.: +55 (91) 3271-1726
E-mail:kitamed@hotmail.com
Recebido em / Received / Recibido en: 30/6/2010
Aceito
em / Accepted / Aceito en: 28/9/2010