








Serviços Personalizados
Journal

Artigo
 texto em
texto em  Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Pan-Amazônica de Saúde
versão impressa ISSN 2176-6215versão On-line ISSN 2176-6223
Rev Pan-Amaz Saude v.1 n.3 Ananindeua set. 2010
http://dx.doi.org/10.5123/S2176-62232010000300017
RELATO DE CASO | CASE REPORT | RELATO DE CASO
Relatório de caso: doença de Niemann-Pick com manifestações de insuficiência hepática
Case report: Niemann-Pick disease with liver failure manifestations
Informe de caso: enfermedad de Niemann-Pick con manifestaciones de insuficiencia hepática
Ivanete do Socorro Abraçado Amaral; Lizomar de Jesus Maués Pereira Moia; Erica Furtado Azevedo Coelho; Zilene Lameira de Medeiros; Maria de Fátima Pombo Montoril; Marialva Tereza Ferreira Araujo
Fundação Santa Casa de Misericórdia do Pará, Belém, Pará, Brasil
Endereço para correspondência
Correspondence
Dirección para correspondencia
RESUMO
OBJETIVO: Relatar um caso de paciente portadora de doença de Niemann-Pick que evoluiu com insuficiência hepática crônica, e, desse modo, chamar atenção para esta entidade nosológica.
RELATO DE CASO: Paciente mulher, 34 anos de idade, apresentando esplenomegalia, encaminhada ao hospital da Fundação Santa Casa de Misericórdia do Pará, na Cidade de Belém, Estado do Pará, Brasil, com o diagnóstico de doença de Niemann-Pick, confirmada por dosagens enzimáticas para investigação de doença hepática. Paciente apresentava cirrose que evoluiu cinco anos depois com insuficiência hepática crônica descompensada, tendo sido encaminhada para transplante de fígado.
CONSIDERAÇÕES FINAIS: Assinala-se que esta doença, apesar de rara, pode ser uma das causas de doença hepática crônica, e deve ser diagnosticada precocemente, visto que a expectativa de vida do paciente depende da importância e repercussão dos sintomas.
Palavras-chave: Doenças de Niemann-Pick; Esfingomielina Fosfodiesterase; Esplenomegalia.
ABSTRACT
OBJECTIVE: To report the case of a patient with Niemann-Pick disease who developed chronic liver failure, and to thus draw attention to this nosological entity.
CASE REPORT: A female patient, 34 years of age, presented with splenomegaly and was referred to the Fundação Santa Casa de Misericórdia do Pará hospital, in the City of Belém, Pará State, Brazil, with a diagnosis of Niemann-Pick disease confirmed by enzymatic dosages for investigation of her liver disease. The patient developed cirrhosis after five years with decompensated chronic liver failure and was referred for liver transplantation.
FINAL CONSIDERATIONS: It is noted that this disease, although rare, can be a cause of chronic liver disease and should be diagnosed early, as the patient's life expectancy depends on the magnitude and impact of the symptoms.
Keywords: Niemann-Pick Diseases; Sphingomyelin Phosphodiesterase; Splenomegaly.
RESUMEN
OBJETIVO: Relatar un caso de paciente portadora de la enfermedad de Niemann-Pick que evolucionó con insuficiencia hepática crónica, y, de ese modo, llamar la atención para esta entidad nosológica.
RELATO DE CASO: Paciente mujer, 34 años de edad, presentando esplenomegalia, encaminada al hospital de Fundação Santa Casa de Misericórdia do Pará, en la Ciudad de Belém, Estado de Pará, Brasil, con diagnóstico de enfermedad de Niemann-Pick, confirmada por dosaje enzimático para investigación de enfermedad hepática. La paciente presentaba cirrosis que evolucionó cinco años después hacia insuficiencia hepática crónica descompensada, habiendo sido encaminada para transplante de hígado.
CONSIDERACIONES FINALES: Se destaca que esta enfermedad, a pesar de rara, puede ser una de las causas de la enfermedad hepática crónica, y debe ser diagnosticada precozmente, visto que la expectativa de vida del paciente depende de la importancia y repercusión de los síntomas.
Palabras clave: Enfermedades de Niemann-Pick; Esfingomielina Fosfodiesterasa; Esplenomegalia.
INTRODUÇÃO
A doença de Niemann-Pick (DNP) é uma doença rara, lisossômica ou de acumulação, em que a deficiência de uma enzima específica tem como resultado a acumulação de esfingomielina, um produto do metabolismo das gorduras, sobretudo quanto a fibroblastos. É considerada uma doença generalizada e grave. Esta doença apresenta atualmente seis subtipos, dependendo da gravidade da deficiência enzimática: subtipo A, com forma neuropática aguda; subtipo B, a forma visceral; subtipo C, forma neuropática crônica; subtipo D, variante nova escocesa; subtipo E, forma adulta; e subtipo F, doença do histiócito azul-mar1,2. Em sua forma juvenil grave, a enzima está completamente ausente, o que leva ao desenvolvimento de alterações graves no sistema nervoso, em razão do nervo não conseguir utilizar a esfingomielina para produzir a mielina necessária para as bainhas que, normalmente, envolvem muitos nervos3. Estudo observou que a distribuição geográfica do subtipo B tem maior incidência na França (Europa), Estados Unidos e Canadá (América do Norte), Brasil (América do Sul) e Japão (Ásia)4.
A DNP é diagnosticada por meio de anamnese e exame físico detalhado, apoiados pela biópsia de medula óssea com achados de células de Niemann-Pick característicos, os histiócitos azul-mar. Nos Estados Unidos, o subtipo A ocorre com maior frequência, abrangendo cerca de 85% dos casos desta doença; e estima-se que o subtipo C afete cerca de 500 crianças. A DNP atinge recém-nascidos, crianças e adultos de ambos os sexos. O subtipo A se manifesta nos primeiros meses de vida e o paciente morre na infância; no subtipo B, os primeiros sintomas aparecem na infância, de forma não neuropática, porém muitos sobrevivem até a idade adulta; e os subtipos C e D apresentam distúrbios neurodegenerativos, com início nos primeiros 2 anos ou mais de vida, ou, ainda, no final da infância5,6.
OBJETIVO
Relatar um caso clínico de paciente com DNP que evoluiu com insuficiência hepática crônica descompensada.
RELATO DO CASO
Mulher solteira, com atividade restrita ao lar, escolaridade ao nível do ensino fundamental incompleto, natural do Município do Acará, procedente de Santa Izabel do Pará, com 26 anos de idade quando foi atendida pela primeira vez no ambulatório de fígado do hospital da Fundação Santa Casa de Misericórdia do Pará (FSCMP), na Cidade de Belém, Estado do Pará, Brasil, em setembro de 2002, encaminhada pela Fundação Centro de Hemoterapia e Hematologia do Pará (HEMOPA) com o diagnóstico de DNP. Apresentava dosagem de esfingomielinase de leucócitos e fibroblastos diminuídos: 0,12 nanomoles/h/mg/proteína (0,74 a 4,91) e 1,9 nanomoles/h/mg/proteína (42 a 72), respectivamente. Foram realizadas no HEMOPA as dosagens de β-glicosidase e quitotriosidase, as quais foram normais, e o mielograma, cujo resultado mostrou: presença de grande número de macrófagos intersticiais na série monoplasmocitária, compatível com síndrome do histiócito azul marinho ou DNP.
Neste período inicial, chamavam a atenção a esplenomegalia (baço tipo IV) e os marcadores sorológicos para os vírus das hepatites B e C, que eram negativos. Na primeira consulta, apresentava turgescência jugular, hepatoesplenomegalia, máculas hipocrômicas e hipercrômicas de limites imprecisos nos membros inferiores. Na anamnese dos antecedentes mórbidos pessoais (AMP), a mãe da paciente referiu que, desde os 3 anos de vida, a paciente apresentava um abdome aumentado e, na dos antecedentes mórbidos familiares (AMF), a paciente informou que tem cinco irmãos saudáveis. Com relação aos hábitos, negou uso de álcool, tabagismo ou uso de drogas ilícitas.
Na primeira consulta, em 2002, no exame físico, a paciente se mostrou consciente, orientada no tempo e no espaço, com pressão arterial 140x80 mmHg, frequência cardíaca de 90 batimentos por minuto, icterícia (++/+4), manchas hipercrômicas e hipocrômicas nos membros inferiores, emagrecida, com altura de 1,49 m, peso de 45 kg, baço tipo IV e fígado a 2 cm do rebordo costal direito. Fez exame neurológico, com resultado normal. Neste período, apresentava tempo da atividade da protrombina (TAP) 100% quando foi submetida á biópsia de fígado na FSCMP; e o estudo anatomopatológico, feito pelo Departamento de Patologia da Universidade Federal do Pará, evidenciou DNP pela presença de células esponjosas, além de cirrose. A endoscopia digestiva alta (EDA) não revelou varizes esofágicas. Os exames de 3 laboratório resultaram em: hemácias: 3,9 milhões/mm3, Hb 9,8g%, contagem de plaquetas 160.000/mm3, leucócitos 6.100/mm3, ureia 33 mg/dL, creatinina 0,9 mg/dL, GGT 65 U/mL, glicose 95 mg/dL, proteínas totais 7,6, albumina 4,0, globulina 3,6, AST 92 U/mL, ALT 67 U/mL, alfafetoproteína 2 ng/mL. Na densitometria óssea, demonstrou osteoporose de coluna lombar e osteopenia de colo do fêmur, tendo sido prescrito alendronato de sódio 70 mg semanal, cálcio e vitamina D diariamente.
A paciente deixou de comparecer ao ambulatório e, ao retornar, em 2005, apresentava baço ao nível da crista ilíaca esquerda, hemácias 4,6 milhões/mm3 Hb 11,5 g/dL, Htc 36%, plaquetas 113.000/mm3, leucócitos 4.600/mm3, TAP 75%, TP 14", INR 1.1, creatinina 0,5, ferro sérico 60, FA 76, GGT 55, glicose 90, TGO 61, TGP 25. Na EDA a paciente apresentou um cordão varicoso de pequeno calibre, sendo-lhe prescrito propranolol.
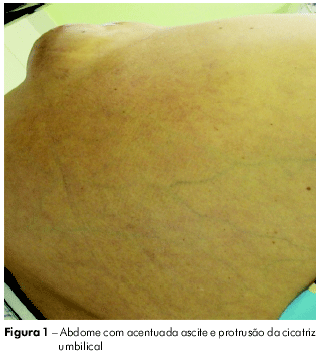
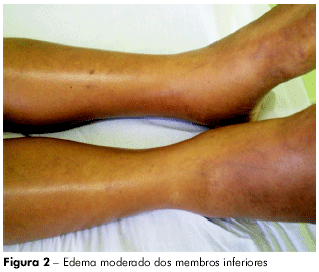
A paciente se ausentou por mais algum tempo, retornando com descompensação do quadro: com ascite (Figura 1) e emagrecimento, tendo sido internada por várias vezes. Em maio de 2009, quando se internou no hospital da FSCMP, apresentava ascite de grande volume, circulação colateral, edema de membros inferiores (Figura 2) e desconforto respiratório; fazia uso de espironolactona, propranolol, lactulona e vitamina K. Aos exames, apontou: leucócitos 3,2, Hb 11,8, glicose 97, plaquetas 77.000, BT 1.94, BD 0,5, BI 1.42, FA 75, GGT 100, creatinina 0,70, ureia 29, proteínas totais 6,2, albumina 2,6, globulina 3,6, Na 140, K 3,5, AST 14, ALT 50 , TAP 24", AP 30,2%, INR 2,07, MELD = 17. Foi submetida à tomografia de abdome, em junho de 2009, que indicou volumosa ascite, fígado pouco aumentado à custa de lobo hepático esquerdo com contornos lobulados, apresentando parênquima com densidade finamente heterogênea; baço acentuadamente aumentado de volume, com densidade tomográfica homogênea e imagem sugestiva de circulação colateral entre o baço e o rim. Tomografia de tórax, realizada em junho de 2009, mostrou pulmões aumentados de volume, sinalizando áreas com diferença de atenuação.
A paciente recebeu alta hospitalar com melhora do quadro clínico e foi encaminhada para acompanhamento clínico no ambulatório de fígado da FSCMP, e, deste, foi encaminhada para programação de transplante de fígado. Não foi observado envolvimento neurológico, tendo sido sugerida DNP subtipo B.
DISCUSSÃO
A DNP subtipo B no adulto é rara, afeta igualmente ambos os sexos, pode ocorrer em todas as populações com incidência de 1 para 250 mil pessoas. Os indivíduos afetados por este subtipo, em sua maioria, atingem a idade adulta sem comprometimento neurológico ou da capacidade intelectual5,7. Essas descrições evidenciam a raridade do caso relatado.
O subtipo B é uma doença autossômica recessiva, resultante da mutação no gene SPDM1, presente no cromossomo 11 (p15,1 a p15,4) cromossomo 11p15, 1 a p15, 48, responsável pela deficiência da esfingomielinase ácida, porém com acúmulo de esfingomielina somente no sistema dos monócitos macrófagos, e, devido ao fenótipo (subtipo B), ocorrem sintomas viscerais5,1. No presente caso, os exames de ensaio enzimático individualizaram redução da enzima em questão, com sintomas viscerais evidentes na paciente. Dentre as manifestações mais frequentes da DNP subtipo B, foram encontrados baixa estatura, hiperesplenismo, pancitopenia e alterações de função hepática6,9.
A presença de ascite de moderada intensidade, circulação colateral, distúrbio de coagulação e a presença de cordão varicoso em esôfago são sinais da cirrose hepática e de gravidade na DPN do subtipo B5. A esplenomegalia e/ou hepatomegalia, nesse subtipo, costuma ser detectada nos primeiros anos de vida, durante exame físico de rotina; entretanto, nos casos muito leves, o aumento é sutil, e, por conseguinte, só é perceptível na adolescência ou na vida adulta6. A paciente em questão teve diagnóstico tardio, aos 25 anos de idade, embora a mãe referisse que desde a infância já observara aumento de volume de abdome.
CONSIDERAÇÕES FINAIS
O caso relatado se enquadrou na literatura como a DNP subtipo B no que tange aos achados clínicos, laboratoriais e histopatológicos. Ressaltamos a importância dessa enfermidade, a qual pode ser diagnosticada ainda na infância e pode ter evolução desfavorável, levando o paciente a transplante de fígado.
REFERÊNCIAS
1 Pavlu-Pereira H, Asfaw B, Poupetová H, Ledvinová J, Sikora J, Vanier MT, et al. Acid sphingomyelinase deficiency: phenotype variability with prevalence of intermediate phenotype in a series of twenty-five Czech and Slovak patients. A multi-approach study. J Inherit Metab Dis. 2005;28(2):203-27. DOI:10.1007/s10545-005-5671-5 [ Links ]
2 Garver WS, Francis GA, Jelinek D, Shepherd G, Flynn J, Castro G, et al. The National Niemann-Pick C1 disease database: report of clinical features and health problems. Am J Med Genet A. 2007 Jun;143A(11):1204-11. DOI:10.1002/ajmg.a.31735 [ Links ]
3 Spence M, Callahan J. Sphingomyelin-cholesterol lipidoses: the Niemann-Pick group of diseases. New York: McGraw-Hill; 1989.
4 Calogera MS, Desnick RJ, McGovern MM, Wasserstein MP, Schuchman EH. The demographics and distribution of type B Niemann-Pick disease: novel mutations lead to new genotype/phenotype correlations. Am J Hum Genet. 2002 Dec;71(6):1413-9. [ Links ]
5 Schwartz RAl. Niemann-Pick disease. New Jersey: Medical School; 2008.
6 Goldman L, Ausiello D. Tratado de medicina interna. 21. ed. Rio de Janeiro: Guanabara Koogan; 2001. 1231 p.
7 Schuchman EH, Desnick RJ. Niemann-Pick disease types A and B: acid sphingomyelinase deficiencies. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill; 2001. p. 3589-610.
8 Takahashi T, Suchi M, Desnick RJ, Takada G, Schuchman EH. Identification and expression of five mutations in the human acid sphingomyelinase gene causing types A and B Niemann-Pick disease: molecular evidence for genetic heterogeneity in the neuronopathic and non-neuronopathic forms. J Biol Chem. 1992 Jun;267(18):12552-8. [ Links ]
9 Wasserstein MP, Desnick RJ, Schuchman EH, Hossain S, Wallenstein S, Lamm C, et al. The natural history of type B Niemann-Pick disease: results from a 10-year longitudinal study. Pediatrics. 2004 Dec;114(6):672-7. DOI:10.1542/peds.2004-0887 [ Links ]
 Correspondência / Correspondence/ Correspondencia:
Correspondência / Correspondence/ Correspondencia:
Erica Furtado Azevedo Coelho
Conjunto Mendara I, rua J, no 110
Tel.: +55 (91) 3271-1726
E-mail:kitamed@hotmail.com
Recebido em / Received / Recibido en: 30/6/2010
Aceito em / Accepted / Aceito en: 28/9/2010