









 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkThe Human Genome Project had identified that we have approximately 22,000-25,000 protein-coding genes annotated in a 3 billion base pair haploid genome, a nearly complete list of the human genes. However, there is a new layer of information beyond the DNA sequence. Epigenetics and epigenomics are upcoming fields that started having an enormous impact on biomedicine. Broadly defined, epigenetics refers to the chromatin-based mechanisms important in the regulation of gene expression that does not involve changes to the DNA sequence, and is related to gene expression1. Epigenomics refers to the constellation of the epigenetic modifications in a cell type at a specific time point2. Recently, epigenetics was considered the epicentre of modern biomedicine because of the study of non-DNA sequence-related heredity that can help to explain the relationship between an individual's genetic background, environment, aging, and disease3. The most studied epigenetic modification is DNA methylation, a covalent addition of a methyl (CH3) group to the nucleotide cytosine. In addition, histone density, post-translational modifications, and RNA-based mechanisms are also included1. The importance of DNA methylation is emphasized by the growing number of human diseases that are known to occur when this epigenetic information is not properly established and/or maintained; and there is increasing interest in the developing ways of pharmacologically reversing these abnormalities, some already approved by the Food and Drug Administration (FDA)2. DNA methylation could be seen in diseases such as cancer, mental retardation, psychiatric diseases, neonatal diseases, Alzheimer's disease, and others2,4. Here, we will summarize the molecular machinery associated with epigenomics, focusing on cancer and oncology (Figure 1).
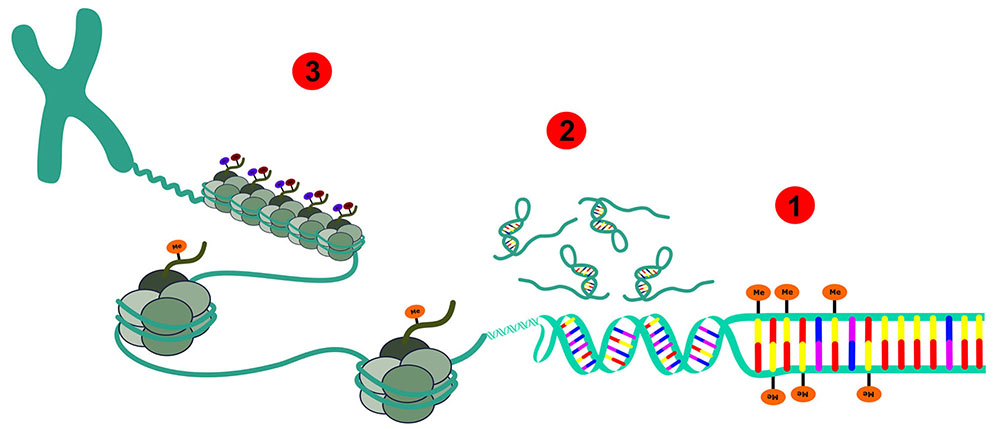
Schematic representation: 1 - DNA methylation; 2 - Non-coding RNAs; 3 - Histone modifications.
Figure 1 - Depiction of all epigenetic modifications that can occur in an eukaryotic epigenome of normal and cancer cells and the chromatin
DNA METHYLATION AND CANCER
The methylation of DNA was discovered by Hotchkiss (1948) in calf thymus and occurs mostly in the fifth carbon of cytosines5. The nucleoside 5-methyldeoxycytidine (5mC) is often considered to be the fifth base of the genome. The distribution of DNA methylation in eukaryotic genome, studied by Sinsheimer in 1955, revealed > 90% of the 5mC methylated in the sequence CpG6. In the early days of epigenetic studies, it was proposed that cytosine methylation in eukaryotic DNA could be a stably inherited modification affecting cellular differentiation. In fact, recently many processes involving DNA methylation have been studied, for example, cell type-specific variation in methylation patterns, involvement of methylation in cellular processes such as gene regulation, DNA protein interactions, cellular differentiation, suppression of transposable elements, embryogenesis, X-inactivation, genomic imprinting, and tumorigenesis2,4. In addition, drugs developed to target the epigenome of cancer cells are already approved by FDA for some types of malignancies2.
Briefly, DNA methylation patterns are erased in the very early embryonic stage and reestablished in every individual at the time of implantation. As a consequence, this allows the embryonic stem cells to differentiate into any possible specialized cell. The data of 5mC from Sinsheimer's report (1955) suggested that > 90% of DNA methylation in calf thymus is determined by the dinucleotide sequence CpG6. The hypothesis that 5mC residues are preferred sites for a spontaneous DNA mutation could partially explain the underrepresentation of the CG dinucleotide (methylated plus nonmethylated forms). In the total DNA and the structural genes of higher eukaryotes, there is a preferential occurrence of 5mC in the highly repetitive noncoding DNA7. The base 5mC is thought to undergo a transition mutation via deamination to produce thymine, which in turn might result in the accumulation of TpG and CpA, with the depletion of CpG, during evolution7.
Deregulation of DNA methylation was already associated with several types of cancer and different stages of carcinogenesis2,4,8. Recently, it was shown that the mammalian genomic DNA contains not only 5-methylcytosine, but also 5-hydroxymethylcytosine (5hmC), which is now considered to be the sixth base of the genome in higher organisms9. Interestingly, hmC was already found in mammalian DNA in 197210, though their presence remained uncertain because the later studies were unable to reproduce the data. There is still limited information about the new sixth base that was first detected in neurons and is not (or poorly) present in the cancer cells lines (e.g., HeLa)9.
HISTONE MODIFICATIONS AND CHROMATIN DYNAMICS IN CANCER
Chromatin, associated with active promoters, is composed of nucleosomes spaced with core histones that are in highly acetylated state11. This situation, along with many other post-translational modifications, provides maximal gene activation and transcription. There are many mechanisms proposed for gene silencing. The second one, working in concert with DNA methylation, are histone modifications also known as the "Histone Code".
The eukaryotic genome is packaged into chromatin, a highly ordered structure that contains DNA, RNA, histones, and other chromosomal proteins. Chromatin was originally classified into two domains, euchromatin and heterochromatin, active and inactive regions, respectively. The major determinants of the chromatin structure are the histone proteins. DNA coils around histones forming nucleosomes, which compact the genome while still allowing access for active processes such as transcription, replication, and DNA repair. Each nucleosome core particle comprises ~ 147 bp of DNA wrapped in 1.7 turns around a protein octamer, formed of two molecules each of the four highly conserved histones H2A, H2B, H3, and H4. The histone domains fold together in antiparallel pairs: H3 with H4 and H2A with H2B12. All four histones also have 'tails' that extend outside of the nucleosome core and are subject to diverse post-translational modifications that are important in gene regulation and chromatin compaction. These covalent modifications, primarily at the N-terminal histone tails, include methylation, acetylation, phosphorylation, ubiquitylation, sumoylation, and ADP-ribosylation. The lysine residues in the histone tails are a common substrate for modifications where acetylation involves small chemical groups, whereas ubiquitylation and sumoylation add large two-thirds the size of the histone proteins themselves. Another degree of complexity is that methylation can occur several times (mono-, di-, or trimethylation)13. Some of the functional outcomes of these modifications are clear, such as acetylation activating, whereas sumoylation seems to be repressing, and these two modifications may mutually interfere. In contrast, methylation and ubiquitylation might have variable effects, depending on the precise residues and context. For example, trimethylation of lysine 4 in histone H3 (H3K4me3) occurs at the 5' ends of genes that become induced, whereas H3K9me3 occurs in compact pericentromeric heterochromatin, which is transcriptionally inactive. Various models such as the charge-neutralization model and the histone code14, as well as a signaling-pathway model are important in this process. Again, deregulation in the mechanisms associated with the so-called "histone code" were already described in different types of cancer.
NON-CODING RNAs AND CANCER
It is clear that non-protein coding RNAs (ncRNAs) dominate the transcriptional output in mammals and other complex organisms. Indeed the eukaryotic genome, rather than being viewed as an island of protein-coding genes in an expanding sea of evolutionary "junk" DNA, may be better thought of as an RNA machine15,16. There are many processes in which ncRNAs (RNAs that are not translated to proteins) have a specific function in the epigenome. In addition, deregulated expression of several of these RNAs are associated with tumorigenesis and cancer development in different types of cancer16.
The ncRNAs can be classified according to their size as small non-coding RNAs (< 200 bp) or long non-coding RNAs (lncRNAs) (> 200 bp)16. MicroRNAs are involved in a number of cellular functions, such as regulation of proliferation, cell differentiation, and apoptosis in both healthy and cancerous cells. Apparently, a network of interactions occurs between the lncRNA and microRNA (miRNA, a type of small non-coding RNA) pathways17. The lncRNAs can act as molecular sponges by capturing the miRNAs, thereby preventing the action of the latter18. The ncRNAs can also act in competition with endogenous RNAs, promoting cell proliferation in prostate cancer19.
Therefore, we believe that, in the near future, ncRNAs could be exploited to identify new cancer biomarkers for disease prediction, progression, and also to develop new therapeutic alternatives to the conventional ones used currently in oncology. Together, all these epigenetic changes are very important to maintain the stability of the so-called Epigenome in normal cells, and problems in these processes are directly linked to cancer development and progression.