








Servicios Personalizados
Revista

Articulo
 Portugués (pdf)
Portugués (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBoletim de Pneumologia Sanitária
versión impresa ISSN 0103-460X
Bol. Pneumol. Sanit. v.9 n.1 Rio de Janeiro jun. 2001
Mecanismo imunitário da tuberculose síntese e atualização
José Rosemberg
Professor Titular de Tuberculose e Pneumologia da Faculdade de Ciências Médicas de Sorocaba da Pontifícia Universidade Católica de São Paulo. Membro do Comitê Técnico- Científico de Assessoramento em Tuberculose do Ministério da Saúde
Introdução
Com a descoberta do bacilo da tuberculose por Koch em 1882, ascenderam-se as esperanças de se imunizar organismos passivamente com a administração de anticorpos extraídos do sangue de animais tuberculizados. A esperança maior foi a de poder curar os pacientes tuberculosos com a soroterapia. É enorme a bibliografia acumulada até aos anos 30 do século passado. Para obtenção de anticorpos usaram-se bacilos tuberculosos atenuados experimentalmente, mortos pelo calor, extratos antigênicos proteínicos, lipídicos, lipoproteínicos e glucídicos, somando cerca de sete dezenas. Diversas micobactérias não tuberculosas (na época chamadas paratuberculosas) foram usadas para ativar a imunidade antituberculosa. Entre elas, a mais difundida foi a vacina de Friedman, que é uma micobactéria isolada da tartaruga. Os anticorpos produzidos pelos organismos receptores desses antígenos são opsoninas, precipitinas, aglutininas e anticorpos fixadores do complemento. Nunca se conseguiu produzir uma bacteriolisina capaz de lisar o Mycobacterium tuberculosis. Esperanças surgiram com a abelha Galleria Melonella, cuja larva alimenta-se com a cera de abelha, produzindo substância destruidora do bacilo de Koch. Em animais inoculados com o Mycobacterium tuberculosis, mesmo saturados desses anticorpos, jamais se conseguiu impedir a evolução generalizada da doença para a morte em poucos dias.
Como se vê, demorou para se perceber que a imunidade da tuberculose não tem base humoral,(1,2,3,4,5).
Ironicamente foi um novato em pesquisas, Madelaine 1914(6) que com tese de doutoramento e procedimento denominado "derivação dos leucócitos", foi um dos primeiros a demonstrar de maneira engenhosa o papel protetor das células mononucleadas contra o Mycobacterium tuberculosis. A inoculação deste no peritônio de animais provocou a chegada de leucócitos e ulteriormente de células mononucleares (monócitos e macrófagos). Os germes foram primeiramente fagocitados pelos leucócitos. Estes não conseguiram destruir os bacilos, que foram levados pela circulação para vários órgãos, produzindo lesões generalizadas. Outros animais receberam, por via endovena esporos de aspergilos mortos pelo calor. Meia hora depois, bacilos foram introduzidos no peritônio. Os germes foram fagocitados, destruídos e/ou detidos no local pelas células monocitárias, enquanto os leucócitos encontravam-se no sistema venoso, entretidos em fagocitar os aspergilos. Nos anos subseqüentes, acumularam-se as comprovações do papel dos macrófagos e monócitos em geral, na imunidade contra a tuberculose. Os estudos de Lurie e colaboradores são os mais abrangentes e clássicos. Demonstraram que os macrófagos de organismos imunizados contra a tuberculose exercem a defesa contra os bacilos de Koch, sem a participação de qualquer espécie de anticorpo. Na câmara anterior de um dos olhos de coelho normal, introduziram-se macrófagos oriundos de animal também normal, e no outro olho, macrófagos de animal intensamente imunizado contra a tuberculose. Em ambas as câmaras introduziram-se bacilos virulentos. Na primeira câmara, os germes multiplicaram-se normalmente, ao passo que na segunda houve nítida inibição, sendo os bacilos destruídos em sua quase totalidade. Esse mesmo procedimento efetuou-se com soro de animal normal e soro rico de anticorpos de animal fortemente imunizado. Não se verificou qualquer efeito sobre os germes. Quando os anticorpos foram associados aos macrófagos normais ou imunizados, a ação destes em nada alterou-se. No correr do tempo, múltiplos estudos confirmaram plenamente que a imunidade na tuberculose é essencialmente mediada por células, não tendo base humoral e que os anticorpos constituem apenas epifenomeno do processo imunitário (7,8,9,10,11).
O fato de a imunidade antituberculose ser mediada por células atrasou por quase um século o seu melhor conhecimento, pela demora de técnicas adequadas para a cultura de células e para sua manipulação. Só com o surgimento da biologia molecular tornou-se possível o alargamento das pesquisas e o aprofundamento dos conhecimentos sobre a complexidade do processo imunitário na tuberculose.
Mecanismo imunitário da tuberculose
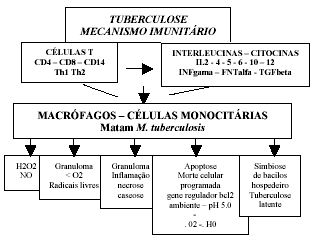
A imunidade antituberculose não resulta de ação específica direta e precisa contra o Mycobacterium tuberculosis. O ataque a este constitui processo de grande complexidade, variando sob ação de circunstâncias diferentes, nem todas bem definidas e com intervenção de elementos múltiplos, razão por que é mais adequado denominá-lo "mecanismo imunitário". Na literatura de língua inglesa emprega-se correntemente a expressão resistência adquirida.
O mecanismo imunitário na tuberculose encerra grande complexidade de interações celulares, notadamente das células especializadas, englobando o sistema macrófago-célula T. O macrófago alveolar talvez seja a principal célula de defesa. Os linfócitos T mantém a memória da imunidade reagindo rapidamente ante novo ataque da micobactéria. A memória imunitária da célula T se esvai com o tempo, sendo geralmente mantida com novas infecções micobacterianas.
As células do citado sistema não atuam propriamente de forma direta, mas através de moléculas, algumas de natureza hormonal, geralmente com 8 a 14 kDa, denominadas linfocinas, interleucinas ou citocinas; algumas podem se estender a outros processos não imunitários, constituindo as quimiocinas. As linfocinas não têm função imunológica direta, pois atuam através o macrófago. Elas atraem os macrófagos ao local da implantação da micobactéria e os ativam para matar o M. tuberculosis.
Os leucócitos polimorfonucleares têm grande capacidade de fagocitar o Mycobacterium tuberculosis, porém sem nenhum poder de destruí-lo. Se não são englobados pelos macrófagos, exercem papel prejudicial, pois as micobactérias são transportadas para diversos sítios do organismo, lembrando a figura do cavalo de Tróia da mitologia grega.
Em síntese, as principais premissas do mecanismo imunitário contra a tuberculose são as seguintes: (12,13,14,15,16,17 ).
- A imunidade contra a tuberculose é mediada por células.
- Os macrófagos previamente ativados matam os bacilos.
- Antígenos do Mycobacterium tuberculosis ativam linfáticos T para elaborar linfocinas.
- As linfocinas atraem os macrófagos ao sítio da infecção e os ativam para matarem as micobactérias.
- A imunidade contra a tuberculose pode ser suprimida pela depleção das células T e restaurada por agentes que recompõem essas células.
- A imunidade contra a tuberculose pode ser transferida experimentalmente de organismo para organismo, transplantando células T .
- A memória imunitária das células T se esvanece com o tempo e, nos casos de sua transferência passiva, ela perdura durante o tempo em que permanecem vivas.
Nos últimos 25 anos, com o auxílio de técnicas moleculares, operaram-se significativos avanços no conhecimento do complexo mecanismo imunitário da tuberculose. Todavia há fundadas razões para crer que ainda não estão esclarecidos todos os meandros desse processo. As funções imunitárias das células monocitárias e células T são mais conhecidas, porém não estão completamente estabelecidas suas interelações através das citocinas, ainda nem todas conhecidas. Na atualidade passam de 40, tendo sido reclassificadas recentemente( 18 ).
As células T são elaboradas no timo, sendo as CD4 e CD8 as mais potentes na ativação dos macrófagos para matar o Mycobacterium tuberculosis. Elas têm a destacada propriedade de reconhecer os antígenos e outros constituintes do bacilo, para elaborarem as citocinas que ativam os macrófagos(178,179). Essas condições desaparecem totalmente na síndrome de Di George, que é a ausência congênita do timo e tireóides(19). Importantes sub-grupos das células T são as células Th1 (helper) e Th2 (citolóxico), ambas com importantes funções imunológicas(19,24). As primeiras têm significativo papel no complexo imunitário por contribuir na elaboração da interleucina IL2 e de interferon gama (FNIy), os mais atuantes ativadores dos macrófagos (83, 122). As segundas, além de elaborarem interleucinas, participam da lise de células monocitárias que fagocitaram micobactérias: elas também participam da elaboração de anticorpos, receptores pelas células B, que entretanto não possuem função na proteção contra a tuberculose(7, 83, 124).
Nos infectados com o Mycobacterium tuberculosis, é grande o recrutamento das células CD4, sendo identificadas em número elevado no líquido de lavagem broncoalveolar, de pacientes tuberculosos(23). Estas células assim como CD8 são capazes, em certas condições, junto com as Th2 de lisar macrófagos com micobactérias fagocitadas(25,92). As células CD8 também são detectadas no líquido de lavagem broncoalveolar e surgem no sangue periférico de pacientes tuberculosos, sendo seu número associado à extensão das lesões (51,52,53,125).
Identificam-se também células CD14 e CD30, com alto poder defensivo contra o bacilo tuberculoso, pela sua capacidade de também ativar macrófagos(19) .
As principais interleucinas elaboradas pelas células CD4, Th1 e Th2 são (19,21,25,83,122):
- IL2 - a mais potente citocina ativadora de macrófagos e por sua vez também recrutadora das próprias células T. Age em associação com FNIy;
- IL4 e IL5 - ativam macrófagos e participam da ação contra micobactérias em geral;
- IL6 - participa na superação da fase aguda da infecção tuberculosa, geralmente em associação com o fator de necrose tumoral alfa (TNFa);
- IL8 - atrai células T para o local da infecção e mobiliza leucócitos;
- IL10 - aumenta a produção de células T;
- IL12 - regula recrutamento de células T, que por sua vez, é regulada por IL4(20) ;
- NFTa - Fator de necrose tumoral alfa. Participa da formação do granuloma. Regula processo inflamatório e estimula macrófagos a impedir a multiplicação do M. tuberculosis (26,27). Ratos geneticamente deficientes de produzir FNT alfa, IL6 e IL12 são altamente susceptíveis a infecção tuberculosa;
- FNIy - Interferon gama. Participa intensamente na proteção contra o M . tuberculosis, importante elemento na defesa contra micobactérias em geral(28,29). Interferon gama é encontrado no sangue circulante de pacientes tuberculosos em associação inversa com o grau de severidade das lesões na tuberculose experimental em animais e em humanos(29,30,83). Essa molécula tem relevante cooperação no tratamento da tuberculose, acelerando em certos casos as respostas clínicas favoráveis e é bom adjuvante no tratamento da tuberculose multirresistente (31,32,33). Ela também impede a dispersão dos bacilos fagocitados pelos leucócitos polimorfonucleares e participa na formação do granuloma; é potente ativador das células Th1 e Th2,(19,21). Animais com falta de interferon gama por mutação do gene codificador sofrem facilmente processos disseminados pelo M. tuberculosis, BCG e outras micobactérias(21,34,35). Registram-se casos de crianças com defeito genético na produção de interferon gama que vacinadas com BCG sofreram generalização grave da vacina, sendo uma com êxito fatal,(36). Há registro de 4 crianças irmãs com defeito familiar congênito com ausência de produção de interferon gama que tiveram disseminações graves com micobactérias não tuberculosas, tomando praticamente todos os órgãos. Apesar da quimioterapia 3 faleceram(37).
Entre os linfócitos participantes do processo imunitário antituberculose, incluem-se as células gama - delta, cujo citoplasma é rico em granulações e têm a peculiaridade de desenvolver dendritos(19). No sangue circulante, constituem menos de 10% do total das células T(38). Em animais inoculados com o bacilo de Koch, as células gama - delta aumentam rapidamente no sistema linfático e no início da tuberculose em humanos atinge logo, a 25% a 30% do total das células T (39,83). Essas células parecem exercer papel relevante na defesa inicial contra a tuberculose. Também em indivíduos tuberculino - positivos, contatos de tuberculosos, seu número eleva-se significantemente como se fossem guardiãs do processo de infecção pelo M. tuberculosis(40). Há evidência de que essas células colaboram para os estados de tuberculose latente, contribuindo para as defesas garantidoras da simbiose bacilo - organismo(41). Um dos ângulos interessantes do processo imunitário na tuberculose prende-se ao fato de que elementos do corpo bacilar ou do genoma do M. tuberculosis têm a capacidade de mobilizar, recrutar as células T e de intensificar suas ações de defesa quer diretamente e/ou através as citocinas. Dois exemplos devem ser destacados. A glicoproteína lipoarabinomanan da membrana cero-lipídica, envolvente do M. tuberculosis, mobiliza o sistema de células T e ativa a produção do fator de necrose tumoral e interferon gama, que por sua vez ativam o macrofágo para destruir a micobactéria(20,21,22,42,83,123). No DNA do bacilo há uma proteína de kDa6, denominada ESAT-6, que é um dos mais potentes mobilizadores das células imunitárias, agindo em associação com interferon gama(43) (Ver adiante).
Outro ângulo a aumentar a complexidade do processo imunitário da tuberculose decorre do fato de que as células T são ambivalentes, no sentido de que reagem a vários componentes do M. tuberculosis, estabelecendo estados de defesa favoráveis ao hospedeiro, como também reagindo a certas proteínas do germe, estabelecendo estados de hipersensibilidade (medidos pelas respostas à tuberculina) que são prejudiciais, porque condicionam o agravamento da infecção tuberculosa. Esse aspecto da tisiogênese foi sintetizado por Rich(3) há tempos com sua então famosa equação, na qual a lesão é igual a fatores desfavoráveis colocados no numerador, que são: número de bacilos, mais sua virulência, mais hipersensibilidade; no denominador, estão os fatores favoráveis: imunidade adquirida e resistência natural. O risco de surgir tuberculose-doença depende do predomínio do numerador sobre o denominador, havendo evolução favorável quando este predomina. Esse binômio imunidade-hipersensibilidade governa a evolução da infecção tuberculosa. Há farta evidência acumulada de que, nos infectados com o M. tuberculosis, o risco de desenvolver doença é maior nos reatores fortes à tuberculina, e tanto maior quanto mais intensa é a reação. Inclusive as chamadas reações paradoxais de agravamento da lesões tuberculosas nos co-infectados com HIV, que se vem registrando na quimioterapia, é debitada à exacerbação da hipersensibilidade (44,45).
É intrigante que a mesma célula T seja responsável por dois processos antagônicos, como imunidade e hipersensibilidade. Mais intrigante é que essas duas condições são independentes e dissociáveis. Os componentes do M. tuberculosis que provocam os dois estados são diferentes. Experimentalmente pode-se criar hipersensibilidade sem imunidade e vice-versa. Uma delas pode-se esvair espontaneamente, ou ser suprimida experimentalmente, mantendo-se a outra (3,46,47,178) .
Essa dissociação da resistência imunitária da hipersensibilidade ficou bem confirmada no estudo prospectivo controlado de vacinação BCG efetuado na Inglaterra, pela constatação de que, com qualquer nível de intensidade da reação tuberculínica e mesmo com ausência de resposta à PPD, os vacinados tiveram o mesmo alto grau de proteção contra a tuberculose(44,201).
Ambos os estados, imunidade e sensibilidade, são mediados pela célula T. Os dois estados podem ser criados passivamente transferindo células de organismo infectado com o M. tuberculosis para animais normais. Estes podem responder com sinais de proteção contra o bacilo de Koch e com respostas positivas à tuberculina. Essa situação é temporária, só perdura enquanto as células transferidas não morrerem, ou não forem eliminadas(3,48,49,50). Ainda não está claro porque as células T, conforme os organismos infectados com o M. tuberculosis, desenvolvem maiores ou menores graus de proteção, maior ou menor sensibilidade tuberculínica. Há evidências de implicações genéticas orgânicas, de um lado e de outro, possivelmente de biodiversidade das micobactérias. Esses aspectos serão abordados adiante.
Do exposto, fica patente o significativo papel das células T no processo imunitário da tuberculose. Fica também muito claro como o vírus HIV/AIDS é o maior agravante da historia da tuberculose. Ele destrói o sistema celular T e a imunidade contra o Mycobacterium tuberculosis assenta-se basicamente nesse sistema.
Cabe ao macrófago, com as armas que lhe fornecem as células T, a tarefa de destruir o Mycobacterium tuberculosis, sendo considerado, por isso, como a célula principal do processo imunitário. O macrófago ativado pelas citocinas, aumenta o diâmetro e o número de vacúolos fundidos com os fagolisosomas distribuídos no citoplasma. No interior, o ambiente é hostil para micobactéria, a começar pelo pH muito baixo (em torno de 4.0 a 5.0 (54) ). Com a interveniência do interferon gama e fator de necrose tumoral, o macrófago ativado adquire a capacidade de produzir peróxido de hidrogênio (H2O2) e óxido nítrico (NO), com os quais consegue matar o Mycobacterium tuberculosis(127). A junção de superóxido desmutase ou de catalase decompõe H2O2 em água, anulando a capacidade de destruir o bacilo(19,55,56). Micobactérias não tuberculosas (atípicas), por produzirem grande quantidade de catalase, dificultam a ação bactericida do macrófago. Aliás, há cepas do Mycobacterium tuberculosis, que se defendem contra o peróxido de hidrogênio e o pH baixo do fagolisosoma, aumentando a elaboração de catalase e produzindo amônia para alcalinizar o ambiente(57). Outro mecanismo pelo qual o macrófago mata o bacilo da tuberculose é o aumento da capacidade de sintetizar oxido nítrico (NO). Este tem maior poder bactericida que H2O2(21,58,59). Macrófagos de doentes com tuberculose pulmonar, têm muito maior capacidade de elaborar NO que os recolhidos de pulmões de pessoas sadias: 65% contra menos de 10%(63). Tanto NO, quanto H2O2, atuam da mesma forma, que é a liberação de oxidantes lesivos a micobactéria.
Constatou-se que, sob certas condições, o macrófago pode elaborar citocinas, entre elas, o interferon gama(62). Nem sempre essa célula age sozinha. Pode ter o auxilio de células como CD8 e CD14, fagocitando macrófagos contendo bacilos no seu bojo(25,60). Algumas moléculas, como interferon gama, podem inibir ou anular de todo o fator de crescimento da micobactéria (GEF) e o fator transformador do crescimento (TGF beta)(61). Ao mesmo tempo, identificou-se um fator inibidor do crescimento (GIF), cuja ação pode ser reforçada pelo interferon gama(13).
São, portanto complexas as ações imunitárias na tuberculose e os dados mencionados certamente não esgotam o assunto. Porém precisam ser bem aprofundados, porque governam a tisiogênese. Os principais aspectos são a seguir abordados.
Tubérculo - granuloma tuberculoso
O modelo experimental do tubérculo é conhecido de longa data. No local da entrada do bacilo no pulmão (alvéolo), ocorre um aporte rápido de leucócitos que fagocitam os germes, não conseguem matá-los e podem levá-los a diversos recantos do organismo. Quanto melhor as defesas, chegam mais depressa às células monocitárias para o local do inicio da infecção. Estas podem fagocitar e deter os germes, fagocitados ou não pelos leucócitos. Acorrem células epitelióides por comando de células T. Elas se fundem com células monocitárias, formando os gigantocitos multinucleados - são as células de Langhans. Assim, o fator de necrose tumoral intervém na inflamação aguda, influenciando células monocitárias, transformando-as nos gigantocitos e nas células epiteliais. Formando-se o granuloma, o fator de necrose tumoral participa da evolução para a necrose de caseose no centro. Ao contrário do que pode parecer, esta serve para a contenção dos bacilos, devido ao ambiente anaeróbico que se instala no granuloma como um todo, além do ambiente inóspito nos fagolisosomas. Quando a imunidade se sobrepõe, há evolução favorável do processo. O fator hipersensibilidade torna as células mais sensíveis aos efeitos tóxicos de elementos do corpo bacilar, a maioria de natureza lipoproteínica, entre eles o ácido tuberculoesteárico. A necrose de caseificação amplia-se, aumentando o número de germe livres, extracelulares. A sorte da evolução do granuloma depende do polo que vai predominar. Nas situações de alta imunidade e pouca hipersensibilidade, a necrose não progride, é invadida por fibroblastos, e forma-se um nódulo residual que pode, em certas condições, evoluir para calcificação. Isso porém não impede que dentro do aglomerado cicatricial permaneçam bacilos vivos quiescentes que, por sua vez, manterão a situação com algum grau de hipersensibilidade, fato esse comprovado pela reação à tuberculina que permanece positiva. Nos casos em que a hipersensibilidade se sobrepõe amplamente, a necrose de caseificação extenue-se, o caseo pode liqueficar-se, a proliferação bacilar progride e atingese o estado de tuberculose ativa. Tudo isso sob o comando da hipersensibilidade(64).
Apoptose
Outro caminho da infecção tuberculosa é pela apoptose, em cujo processo os germes morrem juntamente com os macrófagos, esses vitimados pelas suas próprias armas. Apoptose diferencia-se da necrose pelas diferenças morfológicas e bioquímicas na evolução da tisiogênese; os dois processos podem coexistir(65). A apoptose é a morte celular programada. É regulada pelo gene Bcl - 2 que define a sobrevida da célula(66,67) . No fenômeno da apoptose o ato da fagocitose das micobactérias pelo macrófago realiza-se com a participação de interferon gama e de adenosinatrifosfato (ATP). No citoplasma da célula forma-se uma multiplicidade de vacúolos dilatados que englobam os fagolisosomas. O pH baixa rapidamente e há intensa produção de radicais livres, íon superóxido de oxigênio, peróxido de hidrogênio e hidroxila, criando condições inóspitas impedindo a proliferação dos bacilos que morrem rapidamente. O macrófago tem seu metabolismo interrompido, seu DNA fragmenta-se pelas condições desfavoráveis do seu citoplasma e também morre(68,69). Células CD4 e CD8 também participam do processo de apoptose. Os macrófagos recrutados para a apoptose são suicidas, escolhidos para tal fim e já se falou que cumprem missão teleológica(70).
Em síntese, do exposto ressalta-se a alta diversidade das ações intervenientes no processo imunitário contra o Mycobacterium tuberculosis. Do equilíbrio dessas ações com as agressões e defesas do bacilo, pode-se estabelecer um estado de simbiose entre ele e o hospedeiro, caracterizando o estado da infecção latente.
Tuberculose latente
Com o avanço dos conhecimentos dos complicados meandros do mecanismo imunitário da tuberculose, melhor se poderá compreender as condições que facultam ao Mycobacterium tuberculosis conviver com o hospedeiro por decênios, sem morrer, mas sem conseguir multiplicar-se, sem provocar lesões, estabelecendo-se um estado de simbiose.
A tuberculose latente permanece em grande parte como um enigma(128). Cerca de 2 bilhões de pessoas (um terço da humanidade) alberga em seu organismo o bacilo de Koch e desses, apenas 10 % desenvolvem tuberculose ativa. Esse risco é em média 0,2% ao ano, ocorrendo reativação endógena(71,72,73,129). Portanto, imensa parcela da humanidade convive com a micobactéria, sem repercussões sintomáticas.
Uma das dificuldades para explicar esse estado de simbiose, entre o M. tuberculosis e o organismo humano é que não se consegue estabelecer tuberculose latente em animais para servir de modelo e melhor entender o processo. Estudos experimentais com germes atenuados ou com virulentos inoculados em doses mínimas, ou associados com quimioterapia, ou com a administração de corticóides ou outras substâncias, ou ainda analisando a infecção experimental com bacilos em animais com resistência natural (M.tuberculosis em coelhos e outros procedimentos), não trouxeram maiores luzes para entender plenamente a infecção tuberculosa latente(128, 130,131). Experiências com neutralizações do interferon gama e ou do fator de necrose tumoral, ou com imunossupressão por inibição de NOS2, também nada adiantaram(131).
O fato intrigante é que na referida simbiose o bacilo mantém-se vivo sem se multiplicar e a prova de que ele mantém um intercâmbio com o organismo é a permanente resposta positiva à tuberculina. O estado de infecção latente, universalmente chamado tuberculose latente, caracteriza-se por ausência de sinais clínicos. Pesquisas experimentais e histobacteriológicas em humanos demonstram que bacilos de Koch podem manter-se vivos dentro de calcificações e mais freqüentemente em focos granulomatosos com pouco e mesmo sem nenhum oxigênio, impedindo sua multiplicação. Entretanto, o germe é aeróbio. Não havendo novas infecções, a memória imunitária das células T se esvai com o tempo. Devem, portanto, intervir outros fatores que concorrem para manter essa simbiose(73). Para viver em ambiente tão pobre de oxigênio, pode-se pensar que o bacilo esporula. Contudo, nunca se demonstrou de forma convincente que o M. tuberculosis possa desenvolver esporos. Uma explicação sugerida a pouco tempo(74) é de que o germe adapta-se a essas condições desfavoráveis por produzir grandes quantidades de uma proteína alfa cristalina de 16 kDa, quando ele está nessas condições estacionárias. Possivelmente essa proteína forma como que uma camada em torno de seu corpo que lhe garante proteção para reduzir ao máximo seu metabolismo. Além disso, demonstrou-se que o bacilo, em estado de latência, adapta-se a grandes reduções da tensão de oxigênio (pO2), conseguindo permanecer vivo a despeito de viver praticamente enclausurado por muito tempo(75). Por outro lado, o germe em estado de latência pode suprir-se de oxigênio, embora em pequena proporção através o glioxilato(76). O estado de hipóxia pode ativar a enzima glicina-dihidrogenase que libera o glioxilato. Além disso, o bacilo de Koch elabora nitrato-redutase, sendo o maior redutor de nitratos, persistindo nitritos no ambiente da lesão residual. Estes lhe fornecem algum oxigênio que lhe permite sobreviver, embora sem multiplicar-se, em condições de hipoxia dentro da necrose da lesão quiescente(77,78). As células gama-delta parecem contribuir para o estado de latência dos germes(41).
Em suma, para o estabelecimento da tuberculose latente, há pouca probabilidade da intervenção do processo imunitário, parecendo resultar de uma peculiar combinação de eventos, que facultam ao M. tuberculosis a sobrevivência com a níveis metabólicos mínimos. O desequilíbrio desses eventos, quase sempre somados à exaltação da hipersensibilidade, cria condições para o despertar dessa latência do germe, operando-se a chamada exacerbação endógena.
Autores argumentam que a expressão tuberculose latente deveria ser abolida por ser incorreta, porque nos dicionários médicos defini-se infecção latente quando não há sinais nem sintomas, o que se refere ao hospedeiro. A ênfase deve ser sobre o estado fisiológico do germe. Para este também não parece certa a expressão, bacilos dormentes. Segundo esses autores, a denominação "persistência não replicadora" do Mycobacterium tuberculosis, seria mais correta(73,77,78).
Outro significativo aspecto da latência dos bacilos tuberculosos é o estado de persistência com baixíssimo metabolismo que lhes faculta subsistir, não obstante a ação das drogas antituberculosa; estes, quando se reativam, são os maiores responsáveis pelas recaídas pós-quimioterapia. A quimioterapia não consegue extinguir todos os bacilos persistentes dos pacientes tuberculosos, assim como a quimioprofilaxia não mata todos os germes da tuberculose latente. Para os bacilos persistentes da quimioterapia, há os que invocam antigas hipóteses, como a existência de fases de ultravírus, de funções protetoras das granulações de Much, de fases sem a carapaça cerolipídicas eventualmente demonstrável pela microscopia eletrônica. Isso tudo exige confirmação. Não se sabe, inclusive, se as duas situações mencionadas de "persistência não replicadora" são devidas ou não a um mesmo mecanismo(77A).
Se ainda não se conseguiu estabelecer uma ligação da imunidade com a persistência não replicadora da micobactéria, por outro lado sabe-se que a infecção latente é evitada por deficiências imunitárias de origem genética, diminuindo a atividade do sistema células T e macrófagos, com falhas na elaboração de citocinas. Ainda condições genéticas intervenientes no sistema HLA, influem sobre as funções das células T, originando estado de susceptibilidade orgânica para o desenvolvimento de tuberculose ativa(19,83,25,126). Nesses casos, não há condições propiciadoras para estabelecer-se a tuberculose latente. As bases genéticas da proteção contra o M. tuberculosis são expostas adiante. Os diversos elementos e as respostas do complexo imunitário da tuberculose, estão sintetizados no quadro geral.
Importância de teste prático de imunidade na tuberculose
Ainda não se conseguiu um teste demonstrativo da imunidade antituberculosa de fácil aplicação na rotina dos estudos epidemiológicos, como é a prova tuberculínica para detectar os infectados com o Mycobacterium tuberculosis. A razão dessa dificuldade é explicável pelos múltiplos ângulos existentes no mecanismo da imunidade antituberculosa e pela insegurança da sua validade usando-se apenas um deles. Não obstante os percalços, está havendo progresso nesta linha de pesquisa. Foi proposta uma prova de imunidade " in vitro", calcada nos dados fornecidos com a tuberculose experimental em animais(47). A prova proposta e já testada utiliza os passos fundamentais do mecanismo imunitário da tuberculose(13). Macrófagos humanos cultivados são postos em contato com número determinado de bacilos de Koch, juntando-se citocinas elaboradas por células T de organismo infectado com o Mycobacterium tuberculosis. Tudo é colocado em recipiente de cultura, observando-se se os bacilos permanecem vivos, por meio de vários métodos, sendo preferível o radiométrico, e qual o índice do seu crescimento. Pode-se assim apurar se o organismo do qual provieram as citocinas tem ou não imunidade antituberculosa. Evidentemente, este teste de imunidade, já demonstrado como factível, é técnica sofisticada ao alcance de poucos laboratórios, não oferecendo condições operacionais de rotina.
A obtenção de teste simples de imunidade na tuberculose constituirá grande avanço na epidemiologia, na clinica e na vacinação. Estão em curso investigações sobre novas vacinas. Um teste prático de imunidade avaliaria de imediato o poder protetor de uma nova vacina, não havendo necessidade de custosos e longos estudos, como os prospectivos controlados.
Há boas perspectivas para teste de imunidade de aplicação rotineira com os conhecimentos que vêem sendo capitalizados com a decifração do genoma do Mycobacterium tuberculosis.
Relação da biodiversidade do mycobacterium tuberculosis com imunidade e tisiogenease
A imunidade antituberculose tem sido mais investigada em relação ao hospedeiro que ao agente patogênico. Há tempos demonstrou-se que o corpo do bacilo de Koch, independente do fator vida, provoca lesões especificas, pela ação de proteínas, lipoproteínas e lipóides. A introdução de bacilos mortos pelo calor, emulsionados em óleo de vaselina ou parafina, facilita sua dispersão no organismo animal e produz lesões histopatológicas especificas disseminadas, inclusive focos de caseose(78A,78B,78C).
Por outro lado, constatou-se que bacilos de Koch isolados de culturas, inoculados em animais tuberculizados, têm dificuldade de se multiplicar no local da inoculação e nos gânglios satélites devido a imunidade desenvolvida no hospedeiro. Todavia bacilos retirados das lesões do animal e nesse reinoculados multiplicaram-se facilmente. Tudo se passou como se esses germes tivessem adquirido imunidade contra as defesas do animal(79). Essas e poucas outras pesquisas na mesma linha, não tiveram maiores repercussões. Desde logo, porém, as mudanças do biocomportamento do Mycobacterium tuberculosis foram corretamente atribuídas à mutações e a outras alterações genéticas. Algumas cepas foram estudadas por dezenas de anos. Em 1891, no laboratório Saranac, isolou-se um bacilo de criança com curso fatal de miliar tuberculosa. Essa cepa foi denominada R1. Após dois anos de cultura, R1 perdeu a virulência para cobaias, sendo que em altas doses repetidas pode causar focos de caseose. Essa cepa, cuja história foi relatada com quarenta anos de manutenção em cultura(80) , assim permanece até a atualidade. Nesse mesmo laboratório, em 1905, isolou-se um bacilo de paciente com tuberculose pulmonar, batizado de H37. Durante a década dos anos 1920 uma cultura sofreu alterações morfológicas, perdendo a virulência para animais de laboratório. Sua história foi contada também com quarenta anos de seguimento(81). As demais culturas continuam virulentas. Destes germes saíram dois ramos que na atualidade são estudados com métodos moleculares: H37Rv (virulento) e H37Ra (avirulento). Diferenças genéticas estão sendo registradas entre as duas cepas. Estas mostram, em animais, dissemelhança na mobilização de células T e produção de citocinas, alem de diferenças na produção de manose que, junto com arabinomanam, induzem algumas respostas diferentes celulares. Portanto, a resposta do sistema imunitário dos hospedeiros é diversa para H37Rv e H37Ra.(82). A cepa do M. tuberculosis isolada no sul da Índia, denominada variedade Sul - Índia, tem discreta virulência para a cobaia. Nas pessoas infectadas com essa cepa, decorre um tempo longo, inusitado, entre a viragem tuberculínica e as manifestações clinicas da primo-infecção. Há evidência que essa cepa, nos casos que não desenvolvem tuberculose, ao contrario, cria certo grau de resistência contra a doença(82A).
Nos últimos tempos, avolumam-se pesquisas sobre os genotipos das cepas do bacilo da tuberculose. No momento, trabalha-se com três indicadores característicos. Analisa-se o "fingerprinting" da cepa, que é a inserção seqüencial IS 6110 no DNA específica do complexo Mycobacterium tuberculosis. Esta é uma inserção seqüencial com polimorfismo no fragmento de restrição (RFLP - Restriction Fragment Length Polymorphims) que existe no DNA em até 22 cópias dispostas na mesma disposição nas cepas que são do mesmo genotipo. Assim, IS 6110 é como a impressão digital do genotipo do bacilo(82B,82C). Outra forma de análise do genotipo da cepa do bacilo é identificar seqüências de repetição de pares de bases chamadas DR (Direct repeat). Esta técnica denomina-se "spoligotyping"(82C,82D). Outro indicador que está sendo usado é o PGRS, identificando segmentos do DNA da micobactéria ricos em base CG (citosina e guanina)(160 ).
Vem-se observando que cepas com seu determinado genotipo induzem, de alguma forma diferente, o mecanismo imunitário, podendo em graus variáveis dificultar ou alterar a proteção do hospedeiro. Algumas dessas cepas são melhor conhecidas. Nos Estados Unidos, numa pequena área rural, eclodiu um surto agudo de tuberculose, provocando nos infectados intensas reações tuberculínicas e nos doentes lesões com granulomas volumosos e extensa caseificação. A cepa desse M. tuberculosis recebeu a sigla CDC1551. Enquanto a cepa padrão Erdman produz no pulmão do rato 1.000 bacilos em 10 dias e 10.000 em 20 dias, a cepa CDC1551 produz respectivamente 10.000 e 10 milhões de bacilos. Esta revela-se capaz de impedir a proliferação das células Th1(93,94). Outro M. tuberculosis, muito disseminado nos Estados Unidos, cuja sigla é 210, causador de intenso surto muito virulento, tem crescimento rápido dentro do macrófago e influe desfavoravelmente na produção de interleucinas IL6,IL10,Il12 e fator de necrose tumoral alfa(95,96,97).
Desde os primórdios da quimioterapia, constatou-se que cepas do M. tuberculosis resistentes à isoniazida perdem a virulência para a cobaias e camundongos e que cobaias inoculadas com bacilos resistentes à estreptomicina, sobrevivem mais tempo que animais inoculados com germes sensíveis(97A,97B). Entretanto, em ambos os casos, os bacilos permanecem patogênicos para humanos. Registram-se muitos surtos epidêmicos de alta patogenicidade ocasionados por M.tuberculosis multidrogarresistentes e pacientes com esses infectados, sofrem mortalidade elevada, não obstante tratamento com drogas alternativas. Entre essas cepas, inclui-se a cepa W resistente a sete drogas. O estudo do fenótipo dessas cepas, revela que algumas dificultam a mobilização pelo hospedeiro das células do sistema imunitário antituberculoso (90,98,98A,98B,98C,98D,98E,106).
O fenótipo do M.tuberculosis influi nas respostas de defesa do hospedeiro, porque há evidência de gene ou grupo de genes, que contribuem para a mobilização celular e qualidade da imunidade do organismo infectado. As informações sobre esse complexo processo ainda necessitam ser ampliadas e aprofundadas. Já se conhece uma cepa do Mycobacterium tuberculosis, que atenuou sua virulência pela mutação do gene rpoV(97). Sabe-se que no Mycobacterium bovis existe o gene mpb 64 e a proteína ESAT - 6 que não se encontram no DNA do BCG. A perda desses caracteres genéticos e talvez outras mutações não esclarecidas ainda transformavam um bacilo tuberculoso bovino virulento num germe avirulento protetor contra a tuberculose(121).
Em suma, há grandes variações na virulência do Mycobacterium tuberculosis, já conhecidas há muito tempo que é quantitativamente diferente conforme as cepas(90).
O mecanismo imunitário nos infectados com Mycobacterium tuberculosis configura-se de acordo com fatores intervenientes do corpo micobacteriano e do organismo do hospedeiro, que por sua vez é condicionado à fatores genéticos que são abordados a seguir.
Resistência natural e seleção das populações em relação à tuberculose
Bases genéticas
Há evidências sugestivas de diferenças entre os sexos na susceptibilidade de contrair tuberculose ativa, que seria maior nas mulheres, na adolescência, idades jovens, passando a predominar nos homens a partir dos 30 anos, mais ou menos. Fatores econômico-sociais parecem participar dessa diferença, porem com peso muito inferior aos genéticos(98).
Uma das maiores revisões da influência de bases genéticas sobre a evolução da infecção tuberculosa foi levantada com os dados do Royal College of Physicians da Inglaterra, efetuando análise de regressão múltipla para a avaliação das diversas variáveis, além do fator zigótico. Concluiu-se que a concordância para a tuberculose-doença foi significantemente maior entre os gêmeos homozigóticos que nos dizigóticos, evidenciando a importância genética para a tisiogênese(99). Essa conclusão foi confirmada em investigações ulteriores(98).
A relação de fatores raciais com a tuberculose, tem sido mais extensamente investigada na raça negra, sendo as mais relevantes sobre os pretos norte-americanos, abarcando mais de 25.000 indivíduos. A conclusão geral é a que de, mesmo valorizando as piores condições econômico-sociais da raça negra em geral, há evidência segura de sua maior susceptibilidade à tuberculose, em comparação com a raça branca(100,101,102,103). Essa maior susceptibilidade da raça negra para a tuberculose pode ser constatada no mecanismo imunitário, pois o M. tuberculosis prolifera muito mais dentro dos seus macrófagos que nos macrófagos provenientes de brancos. Demonstra-se portanto, que essas células dos pretos têm maior dificuldade de matar os bacilos que as dos brancos(104). Ainda mais, os pretos têm outro fator constitucional agravante para a tuberculose. A vitamina D é um dos estimulantes do processo imunitário contra o M. tuberculosis. Os pretos têm deficiência da vitamina D devido à pigmentação escura da pele que impede sua fotossíntese(105).
Entre os elementos genéticos identificados como fatores de susceptibilidade para a tuberculose, mais extensamente investigados, são os tipos constitucionais HLA (antígeno leucocítico humano). Demonstrou-se incidência de 1,5 a 3,5 vezes maior de tuberculose ativa nos tipos constitucionais HLA - 8B no Canadá(110), no norte da Índia(111), e na Rússia(112). Está demonstrada maior freqüência de tuberculose ativa nos tipos HLA - A11,B8,B15 e DR2(107,108,110). O tipo B15 é encontrado em maior proporção nos negros norte-americanos que, como já mencionado, são mais susceptíveis a desenvolver tuberculosedoença( 109). Entretanto, deve haver situações genéticas que contrabalançam ou anulam o efeito negativo desses tipos HLA para tuberculose porque, por exemplo, entre os chineses de Hong Kong não se encontrou relação estatisticamente significante de tuberculose com tipo HLA susceptível(113).
Partindo dos estudos da resistência natural do rato às infecções de vários agentes parasitários intracelulares e ao Mycobacterium tuberculosis, identificou-se em humanos o gene NRAMP1(114). A resistência natural do rato é regulada pelo gene denominado Bcg ou Lsh ou ainda Ity que possui dois alelos: um dominante Bcg-r que confere resistência à infecção e outro recessivo Bcg-s determinador de susceptibilidade. O Bcg-r intensifica a capacidade dos macrófagos dos órgãos do retículo-endotelial (baço e fígado) de destruir os agentes parasitários fagocitados (115,116). A proteína Nramp1 contida no gene Bcg é considerada como participante da resistência natural do rato (116A,117). A proteína homóloga, identificada no homem foi clonada e grafada MRAMP1. Encontram-se duas mutações dessa MRAMP1 e há evidência de que é muito importante na infecção tuberculosa(118). Embora não se possa garantir a existência em humanos de gene específico governando a susceptibilidade, a proteína MRAMP1 foi encontrada em estudo sobre tuberculose na Gâmbia, oeste da África(119). O polimorfismo desse gene revelou-se significantemente associado à tuberculose. Os indivíduos heterozigóticos para dois MRAMP1, estiveram presentes, em maior proporção, nos casos com tuberculose doença, comparados com o genotipo mais comum; a odd rateio foi 4.07. Concluiu-se que variações genéticas do MRAMP1, estão mais associadas à tuberculose ativa(119). Em Malta, registrou-se que 4 crianças de uma mesma família, sofreram disseminação de micobactéria atípica. Verificou-se que tiveram mutação de MRAMP1 e que a mesma doença havia ocorrido em ancestrais. Pelos dados colhidos, conclui-se que se trata de desordem hereditária autossomica recessiva(120). Vai-se juntando evidência de que a presença de MRAMP1 heterozigóticos, aumenta a susceptibilidade não só para o M. tuberculosis, mas para outras micobactérias(117).
Progressivamente vão-se descortinando os complexos meandros do sistema imunitário na tuberculose inclusive dos fatores genéticos que o governam. Com a decodificação dos genomas humano e do Mycobacterium tuberculosis, espera-se rápido e mais profundo avanço na tisiogênese.
Seleção natural
Estudos moleculares trouxeram à tona informações de que bases genéticas nos humanos são responsáveis pela resistência natural contra o Mycobacterium tuberculosis e de que outras, ao contrário, aumentam a susceptibilidade para contrair a tuberculose. Há consenso de que seres humanos, expostos milenarmente à agressão do bacilo de Koch, foram sendo selecionados, proporcionando coortes relativamente mais resistentes. Desse modo, a maior resistência orgânica humana contra a tuberculose resulta de uma seleção natural que se veio destacando no decorrer dos séculos (132,133,134). Na história recente, há exemplo desse processo, ocorrido na Inglaterra e Suécia. Nas primeiras dezenas dos anos do século 20, quando era alta a prevalência de tuberculose no gado, também era alta a infecção tuberculosa na população, inclusive nas áreas rurais. Anos depois, nessas áreas referidas, observou-se em largas coortes da população adulta associação inversa da mortalidade tuberculosa com a infecção anterior e vice-versa.(135).
A ação secular do agente infeccioso devastando gerações vai selecionando coortes mais resistentes, capazes de se protegerem melhor contra o M.tuberculosis. Essa seleção natural ocorreu na Europa com a expansão da revolução industrial, quando grandes contingentes populacionais foram dizimados pela tuberculose. Em Londres, por exemplo, em meados do século 19, os coeficientes de mortalidade tuberculosa atingiram a 1.100 por 100.000 habitantes. A partir dessa época a epidemia tuberculosa foi decrescendo lentamente, porém acentuadamente. Esse descenso se deveu mais à seleção natural das populações, do que a medidas de saúde pública e conquistas sociais(133, 136 ).
A história mostra que todas as vezes que os colonizadores aportaram nas regiões geográficas dos povos virgens da infecção tuberculosa levaram o bacilo de Koch, vitimando vultosos contingentes dos autóctones, com quadros graves da primo-infecção. Com o correr dos tempos, os aborígenes mais resistentes passaram a influir na epidemia tuberculosa, ocorrendo dominância das formas clínicas crônicas. Isso sucedeu na África, Ásia, Polinésia, Austrália e no continente americano. A dizimação pela tuberculose nos povos conquistados, nos últimos séculos, foi pelo o que hoje está na ordem do dia, o bioterrorismo que os colonizadores empregaram. E não foi tão inconscientemente, pois sociólogos consideraram que "ante a terrível mortandade provocada pela tuberculose nessas populações expostas, o Mycobacterium tuberculosis foi um dos pontos cardeais como maior aliado dos civilizados nas conquistas dos povos aborígenes"(137).
Até a década de 1860 não existia tuberculose na África(138,139). Com as várias colonizações, a doença se disseminou rapidamente nas aglomerações urbanas, com formas clínicas graves, de curso agudo, vitimando grandes contingentes nativos(140,141,142). Em seguida, a onda epidêmica se interiorizou, provocando elevada mortalidade(142, 143,144). No início do século 20, a tuberculose havia atingido quase todo o continente africano, especialmente ao Sul do Sahara e na África do Sul. Mineiros e soldados morriam aos magotes(141,142). Esse quadro também se desenrolou na Ásia, na Indonésia e demais ilhas do Pacífico(143,144). Nos Estados Unidos e Canadá, no final do século 19, os nativos, índios Apaches, entrando em contato com os civilizados, tanto crianças quanto adultos, sofreram a maior dizimação pela tuberculose que se tem notícia, atingindo a mortalidade o incrível coeficiente de 9.000 por 100.000 (145). Aqui no Brasil, na colonização, vieram jesuítas e colonos, muitos deles tuberculosos. Estes contaminaram os índios em massa. Em cartas de Inácio de Loyola e de Anchieta, dirigidas ao Reino, informavam: "os índios ao serem catequizados, adoecem com tosse e febre, muitos cuspindo sangue, a maioria morrendo com deserção das aldeias"(146).
Os pretos, escravos, ao chegarem nos Estados Unidos, contraíam rapidamente a tuberculose, com forma aguda e mortal(147). No Rio de Janeiro, os registros de óbitos da Santa Casa de Misericórdia, no período de 1833 a 1849, por 21 causas ocorridas em escravos, a tuberculose figura com 53,6% do total(148). Durante a primeira guerra mundial (1914-1918), pretos senegaleses foram recrutados pela França para lutarem contra os alemães. Estes contaminaram-se em massa nas trincheiras e foram dizimados com quadros graves de primo-infecção tuberculosa, com múltiplas adenomegalias caseosas, torácicas e abdominais(149). Não há elementos para fixar as características imunobiológicas das gerações que se selecionaram, pela sua resistência natural, contra a tuberculose. Hoje sabe-se que os macrófagos dos negros têm menor capacidade de impedir a multiplicação do M. tuberculosis, que os dos brancos(102,104). Não obstante essa relativa susceptibilidade para com a tuberculose, as formas clínicas que nos tempos iniciais do contato dos pretos norte-americanos, com o bacilo de Koch, eram agudas e disseminadas, são hoje de predominância crônica, mais estabilizadas, como ocorre nos brancos. Isso reflete a seleção natural nos negros, passando a dominar os menos susceptíveis(150). O mesmo ocorreu no Brasil.
A seleção natural preservando as gerações mais resistentes à tuberculose está bem evidenciada na história do povo judaico. Inicialmente, predominava a idéia de que os judeus tinham resistência natural contra a tuberculose mais elevada que as demais etnias. Desde o início do século 20, verificou-se nos Estados Unidos que os judeus imigrados do leste europeu acusavam menor prevalência à tuberculose que a população geral, sendo as formas clínicas mais benignas e crônicas(151). Estudos posteriores, mostraram que essa maior resistência era só dos israelitas da Europa Central que, confinados por séculos nos guetos, casavam entre si. Vitimados em grande proporção pela tuberculose, os mais resistentes deixaram descendentes menos susceptíveis. A crença da resistência natural superior dos judeus foi calcada apenas na observação dos judeus askenases que desenvolveram seleção natural. A contrapartida disso está no fato de que com a fundação do Estado de Israel, os judeus tuberculosos oriundos dos campos de concentração foram para lá levados, ao mesmo tempo que os judeus iemenitas, os quais viveram isolados nos desertos, por milênios, sem tuberculose. Estes, ao entrarem em contato com aqueles, rapidamente contaminaram-se, e muitos desenvolveram primo-infecção grave invasiva do mesmo modo que os negros senegaleses nas trincheiras na França.(132,134,152). Comprovou-se que a superior resistência à tuberculose dos judeus askenases não decorre de característica genética específica e, sim, de uma circunstância histórico-social. Os judeus iemenitas não têm nenhuma resistência natural antituberculosa. Humanos e animais que têm resistência natural à tuberculose possuem macrófagos com maior capacidade de deter e destruir o M.tuberculosis. Não se demonstrou que os macrófagos dos judeus tenham comportamento diferente que as outras etnias na capacidade de matar o M. tuberculosis.
Hipótese aventada recentemente merece ser discutida pela suspeita da possibilidade de uma seleção às avessas, com o advento da era da quimioterapia da tuberculose. Argumenta-se que muitos susceptíveis geneticamente à infecção tuberculosa não estão sendo eliminados; ao contrário, são preservados graças às drogas antituberculosas(136). Se essa hipótese for confirmada, as medidas de controle da tuberculose deverão ser redobradas para eliminar a doença, antes que se avolumem contigentes cada vez mais vulneráveis ao M . tuberculosis.
De qualquer forma, ante os conhecimentos da relativa precariedade do mecanismo imunitário da tuberculose, que não consegue impedir as reativações endógenas, as reinfecções exógenas, e permitindo até que o bacilo conviva em simbiose com o organismo, urge, com os recursos das técnicas moleculares, e a decifração do genoma do M. tuberculosis, criar com auxílio da engenharia genética vacinas mais poderosas capazes de proteger o ser humano contra as deficiências imunitárias apontadas, erigindo-se uma eficiente arma profilática contra a tuberculose.
Vacinas antituberculose - novas vacinas
O único procedimento para criar imunidade antituberculosa nos organismos não infectados e elevá-la nos já infectados é a vacinação. Por enquanto, conta-se apenas com a BCG. Com a engenharia genética, há grandes perspectivas de construir vacinas mais poderosas, como BCG recombinante e vacinas DNA.
Vacina BCG
O BCG deriva de uma cepa do Mycobacterium bovis, que no laboratório do Instituto Pasteur, sofreu mutacionismo, reduzindo sua virulência e mantendo propriedades imunizantes. Entre as alterações do DNA, indentificaram-se a deleção do gene mpb 64 e da proteína ESAT 6, que não existem no genoma do BCG(121). Por sua vez, as cepas de BCG, mantidas em cultura por dezenas de anos em laboratórios dos diversos países, sofreram mutações, variando o poder imunizante e sensibilizante à tuberculina(153,154). Só em 1960(83), esses inconvenientes foram reduzidos em parte, adotando-se o sistema de sementes (seed lot) conservando o BCG, liofilizado, fazendo-se a reconstituição de acordo com as necessidades de consumo(155). Mesmo assim, praticamente não há duas cepas de BCG que sejam inteiramente idênticas geneticamente. As seqüências repetidas no DNA, (DR- direct repeat) que são em torno de 16, sofreram várias deleções, como por exemplo, a cepa trazida para o Brasil que perdeu a DR16(161). Todas perderam a DR1, que se encontra sempre no mesmo locus do DNA do M. tuberculosis, fazendo supor a existência nesse local de gene ou genes ligados á virulência(162). Muitas outras mutações registram-se conforme as diversas cepas(84,85,86). As variações genéticas das cepas espalhadas nos países, explicam as diferenças de mobilização do sistema celular imunitário, portanto do poder de proteção e da hipersensibilidade que cada qual desencadeia (83,87,88,89,90,91,192).
As diferenças dos epitopos do M.tuberculosis são várias. Por exemplo, a inserção seqüencial IS 6110, característica do bacilo, está substituída no DNA do BCG pela IS 987 que difere daquela por dois aminoácidos(156,170).
De qualquer forma, está comprovado que o BCG é o maior indutor de imunidade mediada por células(157). É muito potente a mobilização no hospedeiro das células CD4, CD8, Th1, Th2, macrófagos e células monocitárias em geral do retículo endotelial e a indução de elaboração de citocinas como IL1,IL2,IL4,IL12, interferon gama e fator de necrose tumoral alfa(83,157,159,160). Também são ativadas as células B na produção de anticorpos e receptores, embora não tenham função no mecanismo imunitário da tuberculose(83,157,159,160,161,162).
O BCG produz estado imunitário protetor contra a tuberculose por 10 a 15 anos, porque se mantém vivo dentro dos fagolisosomas dos macrófagos, onde não é destruído como sucede com o Mycobacterium tuberculosis(180) .
O BCG revela o mesmo poder de proteção contra a tuberculose, em indivíduos geneticamente susceptíveis ao M.tuberculosis, por serem fenotipos HLA DR ou por possuírem genes Bcg-s. Isso se verificou em crianças de comunidade asiática susceptível geneticamente, residente na Inglaterra(181).
Entretanto quando o BCG foi submetido a 11 clássicos estudos prospectivos controlados, os índices de proteção antituberculose foram contrastantes. Cinco estudos constataram altos índices de proteção, de 80% para cima: adolescentes de Londres, índios canadenses, índios norte-americanos, jovens de Haiti e crianças de Chicago. Três tiveram resultados medíocres, entre 31% e 34%: jovens de Porto Rico, população de Mandanapalle Índia, e mineiros da África do Sul. Outros três com resultados nulos: adolescentes e crianças da Georgia e de Alabama USA e população de Chingleput, Índia(157). Publicaram-se analises exaustivas desses estudos. Constatou-se que, onde os índices de infecção por micobactérias atípicas não tuberculosas são baixos, insignificantes, a proteção antituberculosa do BCG é alta, ao passo que, onde a prevalência por essas infecções é elevada, os resultados são medíocres ou nulos.
Pesquisas(157,163) em animais infectados com M. avium, ou M. kansasii ou ainda M. fortuitum mostraram que estas desenvolvem certo grau de resistência ao M. tuberculosis. Quando esses animais são então infectados com M. tuberculosis, o BCG tem sua margem de proteção diminuída, porque o nível de proteção que ele confere continua o mesmo que nos animais não infectados por aquelas micobactérias(163). Por isso, os fracos resultados da vacinação BCG nas regiões geográficas com alta prevalência de infecção por micobactérias atípicas, não devem ser debitados à vacina, mas a fatores imuno-epidemiológicos que diminuem o nível de sua ação(163,164,165,166). Há vários fatos intrigantes na interlação imunitária nas diversas espécies de micobactérias, determinando ações aparentemente paradoxais do BCG. Assim, por exemplo, este tem o mesmo poder mobilizador do sistema celular imunitário tanto nos indivíduos das áreas com baixa ou nenhuma infecção de micobactérias atípicas, como nos que vivem em áreas geográficas com alta prevalência dessas micobactérias. Isso foi comprovado em estudo comparativo de mobilização dessas células nos vacinados com BCG, em indivíduos de Chingleput, Índia com alta prevalência de micobactérias atípicas e do bacilo Sul-Índia, e de Londres, onde é negligenciável a freqüência dessas micobactérias(177). Outro fato é que em áreas de alta prevalência de micobactérias atípicas, o BCG não obstante produzir resultados nulos quanto a proteção antituberculosa, exerce proteção significante contra a lepra, como sucede em Malawi(167,168). Por outro lado, o BCG exerce proteção significante contra micobactérias atípicas. Isso foi bem comprovado na Suécia onde um programa de vacinação BCG diminuiu significantemente a incidência de manifestações clinicas em crianças infectadas com o Mycobacterium malmoe (169) .
A maior distribuição das micobactérias atípicas é nas áreas geográficas de baixa latitude, sendo bem menor nas altas latitudes. Naquelas é mais séria a carga da tuberculose. É pois, paradoxal que a vacinação BCG produza menos proteção onde há mais tuberculose e atinja as maiores taxas de proteção nas altas latitudes, onde é baixa a infecção pelo Mycobacterium tuberculosis.
O maior impacto do BCG sobre a epidemiologia da tuberculose, é por diminuir significantemente o risco das manifestações graves hematogênicas da primo-infecção, como a meningoencefalite e a granulia(17). Todas as investigações nesse campo, nos mais diversos países, concordam com esses fatos (171,172,173). Para o cômputo geral da proteção antituberculosa, do BCG, a Escola de Saúde Publica de Harvard, procedeu meta-análise de 1264 estudos cuja metodologia foi considerada correta. A média da proteção antituberculosa em ralação ao risco relativo foi 0.49. (efeito protetor 51%). A média em relação à razão de risco (odd ratio) foi 0.50. (efeito protetor - 50%). Nos estudos que têm registros de óbitos por tuberculose, a proteção em média foi de 71%. Conclui-se, no geral, que o BCG reduz o risco de tuberculose ativa em 50%, nas populações onde é empregado(174).
Há evidência de que existem cepas do M. tuberculosis mais resistentes à imunidade conferida pelo BCG. Com técnicas moleculares constatou-se que em populações onde não se aplicou o BCG sistematicamente, como a Holanda, são muitas as cepas do bacilo de Koch com fenotipos diferentes; ao contrário, nos países com grande cobertura de vacina, são poucas as diversidades de fenotipos da micobactéria. Na região de Beiging, China e na Tunísia, com grande cobertura de BCG, há pouquíssimas cepas de fenótipos diferentes. Isso sugere que se processou seleção natural de cepas mais resistentes que, apesar da vacinação, se impuseram epidemiologicamente(175,176). Conviria averiguar se fato semelhante vem ocorrendo no Brasil, com cobertura vacínica de 70% da infância em 1980 e depois praticamente 100% em muitos Estados.
A proteção do BCG é limitada pelas diferenças no mecanismo imunológico da tuberculose. É por isso que sua proteção contra as manifestações graves da primo-infecção é mais de 80% superior que nas formas pulmonares. Conclusões nessa linha foram extraídas da literatura existente manipuladas pela Escola de Epidemiologia e Medicina Tropical de Londres(173). A forma pulmonar, geralmente mais tardia na historia da evolução da infecção tuberculosa, surge mais freqüentemente pelo esvaimento da imunidade ou, o que parece mais provável, pela intervenção de peculiaridades desfavoráveis dos mecanismo tisiogênicos, como são a exacerbação endógena e reinfecção exógena em organismos já infectados, com estados imunológicos diferentes(173). Deve-se incluir nesse complexo jogo, o papel desfavorável da exacerbação da hipersensibilidade no correr da infecção tuberculosa(157,173).
Há consenso de que o maior poder protetor do BCG é exercido quando introduzido em organismos ainda não infectados por micobactérias. Ele não possui condições biológicas de elevar qualitativa e quantitativamente o mecanismo imunitário nos já infectados com M.tuberculosis, para extinguir a infecção latente (tuberculose latente), prevenir as reativações endógenas e de proteger contra as reinfecções exógenas(157) .
Só vacinas com poder imunológico ampliado para agir positivamente sobre esse complexo quadro poderão exercer impacto significante na epidemiologia da tuberculose. Duas linhas de engenharia genética, com as investigações mais avançadas, são o BCG recombinante e vacinas DNA(188).
BCG recombinante
A imunidade antituberculosa para manter-se necessita que o agente infeccioso ou o BCG permaneçam presentes estimulando o complexo celular imunitário. No caso da infecção pelo M. tuberculosis, havendo esterilização das lesões, a imunidade se esvai completamente. Na vacinação BCG, a imunidade só permanece enquanto o germe-vacina, continua vivo no hospedeiro. E isso ocorre porque ele continua vivo dentro dos fagolisosomas dos macrófagos. É por isso, que o BCG morto tem fraquíssimo poder imunizante. O BCG para manter-se vivo no hospedeiro necessita de um adjuvante que é por ele próprio elaborado(182,183,184,185,186). Há evidência de que uma proteína dimérica de 46 kDa seja elaborada em quantidades diversas, conforme as cepas, variando assim a potência protetora. Portanto é imprescindível contar com BCG vivo e aumentar seu poder protetor. O caminho é inserir no seu DNA epitopos do Mycobacterium tuberculosis e/ou de outras micobactérias. Vários têm sido usados e, com a decifração de genoma do bacilo de Koch, o leque de opções crescerá. Citando apenas os mais estudados, esclareça-se que a técnica de inserir um desses elementos, é complexa, porque nem todas as unidades de BCG os aceitam facilmente através a carapaça cerolipídica e sua inserção do DNA. A operação se faz usando como vetor um plasmídio e micobacteriofago(186,189).
O antígeno Ag 85, extraído do M. tuberculosis, e seus componentes 85A e 85B são bons mobilizadores das células T (190,191,196). ESAT-6 é uma proteína kDa 6, extraído do DNA do M. tuberculosis e ausente no genoma do BCG. Essa proteína é um dos maiores mobilizadores das células especializadas do sistema imunitário da tuberculose. Pela sua alta capacitação de recrutar as células Th1 e Th2, está sendo aproveitada para prova de infecção pelo M. tuberculosis com a vantagem de que as respostas são negativas nos vacinados com BCG. Considerando que a tuberculina - PPD não distingue vacinados e infectados, há boas perspectivas de que ESAT6 se constitua em boa prova para identificação dos infectados com o M. tuberculosis. Essa proteína, pelo seu alto poder recrutador de células imunitárias, é elemento imprescindível para o BCG recombinante (31,187,192,193,194,195). São também investigados, para elevar o poder protetor do BCG recombinante, filtrados de proteínas de choque térmico, como a proteína de 65kDa a lipoproteína A (OspA), todas extraídas do M. tuberculosis (153,197,198,199,200).
Outro ângulo para a fabricação do BCG recombinante é a inserção de epitopos, antígenos dos M. microti e M. vaccae. O Mycobacterium microti foi aplicado também no clássico estudo prospectivo controlado da Inglaterra, produzindo alto grau de proteção antituberculose igual ao conferido pelo BCG(201,202). Entretanto, essa micobactéria não é usada como vacina por provocar extensas lesões necróticas no local de administração. O Mycobacterium vaccae elabora epitopos, proteínas antigênicas, como 19kDa, 45kDa e Ag85, bons mobilizadores das células imunitárias. Há registros de que não desenvolve lesões necróticas e produz baixa hipersensibilidade, reduz a gravidade das lesões tuberculosas, inclusive as cavitárias, quando aplicado associado a quimioterapia ou isoladamente. Genes dessa micobactéria são de interesse para a construção do BCG recombinante(83,203,204,205).
Paralelamente com a elevação do poder protetor antituberculose do BCG recombinante, estuda-se a inserção no seu genoma de proteínas antigênicas e genes de diversos agentes infecciosos, construindo-se uma vacina polivalente. A técnica já está bem definida(186). Genes do Mycobacterium leprae, cujo DNA está quase todo decodificado, estão sendo inseridos no BCG. Do mesmo modo estudam-se genes do Mycobacterium W (não se trata da cepa W do M. tuberculosis multidrogarresistente mencionado no item 4), que se revela com faculdade protetora contra a hanseníase. Assim, pretende-se aumentar o já conhecido poder protetor do BCG contra a lepra,(83,167,186,206,207,208,209,210). É importante o fato de a proteína Nef do vírus HIV, ter sido inserida no DNA do BCG. Ratos imunizados com BCG - Nef tiveram intensa proliferação de células T a qual posteriormente repetiu-se com a introdução da proteína Nef isolada, constituindo a primeira demonstração de resposta especifica contra um antígeno conduzido por BCG recombinante(211). Outros agentes infecciosos estão na pauta das investigações para a construção de um BCG recombinante de ação polivalente.
Ângulo inteiramente diverso da linha das pesquisas citadas é o de capacitar o BCG, mesmo que se extinga sua faculdade de multiplicação dentro dos macrófagos, de continuar como fonte de antígenos indutores, mantendo a continuidade da mobilização do sistema celular de imunidade tuberculose. Isso é conseguido por técnica molecular genética, produzindo o chamado "BCG auxotrofico", criado por mutacionismo com a eliminação do aminoácido leucina, de certo segmento do DNA, que assim impossibilita a produção de enzimas necessárias à sua reprodução. Desse modo o BCG auxotrófico tem sua vida prolongada sem a necessidade da multiplicação nos fagolisosomas dos macrófagos e mantém a continuidade da proteção antituberculosa(157,158,199,212). Outra vantagem do BCG auxotrófico é de não sofrer disseminação generalizada nos vacinados infectados com o HIV e em outros casos de comprometimento da imunocompetência.
Há fundadas razões de manter a expectativa de próxima construção pela engenharia genética, de um BCG de poder ampliado qualitativa e quantitativamente imunizante contra o M. tuberculosis, exercendo significante impacto na epidemiologia da tuberculose; as pesquisas estão na fase 2.
Vacinas DNA
Há mais de 100 vacinas DNA em estudos, diversas na fase 2. Com as vacinas DNA, consignaram-se intensas respostas imunológicas contra a tuberculose, superiores às conferidas com BCG recombinante.
Ao contrário do que se pensava, sua construção é relativamente simples. Genes responsáveis por antígenos bacterianos são inseridos em segmentos de bases nucleotideas do DNA, por meio de plasmídio como vetor(213,214). Entretanto, há uma limitação, que é o desconhecimento de muitos epitopos e antígenos do M. tuberculosis e quais são os mais potentes para imunorespostas(199,215). Todavia, algumas proteínas e antígenos bons indutores das células do sistema imunitário da tuberculose, inclusive já citados na construção do BCG recombinante, surgem como candidatos para a vacina DNA. Entre eles, destacam-se o antígeno Ag85 - 85A, 85B e 85C - antígeno PstS-1, proteínas de choque térmico hsp65 e 70 e ESAT-6(213,216,217,218,219). Já está demonstrado que os antígenos estudados, candidatos à construção de vacina DNA contra a tuberculose, tem elevada capacidade de induzir altos níveis de imunorespostas, mobilizando todo o sistema celular, CD4 - CD8 TH1, TH2, macrófagos, células monocitárias em geral, com elaboração de citocinas mais atuantes, entre estas, o interferon gama e fator de necrose humoral alfa. Ratos que receberam vacina DNA preparada com antígenos do M. tuberculosis, demonstraram essa mobilização celular e adquiriram significante proteção antituberculosa(199,213,216). Com os epitopos acima citados e outros mais, há notícias de experiências em animais com vacinas DNA que sugerem a sua capacidade de impedir exarcebações de focos residuais de tuberculose latente(220).
No estado atual, as vacinas DNA têm o inconveniente do pouco tempo de duração da imunidade. A vacina DNA não é uma célula viva, como o BCG que permanece como fonte constante de indução do mecanismo imunitário celular. Espera-se conseguir o prolongamento de sua ação por meio de adjuvantes, dos quais os mais investigados são os elementos DNT, KLK, cpgs, polyA. Outro procedimento é de incluir a vacina em microesferas(221). Muitas pesquisas estão em curso para a obtenção de vacinas terapêuticas DNA, cuja atuação poderá ser isolada ou associada à quimioterapia. Nessas vacinas, inseriram-se certos carbohidratos, glioxilatos, lipídios, interleucinas IL4, IL12 ou proteínas de choque térmico de 65kDa associados ou não a drogas quimioterápicas(222). Animais tuberculizados que receberam a vacina DNA desenvolveram lesões menos graves. As principais vantagens das vacinas DNA são de impedir reativações endógenas nos organismo com infecção tuberculosa latente e de melhor proteger contra as reinfecções exógenas. Outras vantagens são que não desencadeiam sensibilidade à tuberculina, não necessitam de rede de frio para conservação, sendo prática sua estocagem.
No total, passam de 110 as vacinas antituberculosas em estudos e muitas estão na fase 2.
Novas perspectivas para o controle da tuberculose
São grandes as expectativas de obtenção de vacinas antituberculosas com poder de proteção não só contra a primo-infecção como também contra todas as fases tisiogenéticas dos infectados. Força é reconhecer que no estado atual o armamento profilático antituberculose é relativamente pobre. Grande esperança está sendo depositada na estratégia DOTS recomendada com ênfase pela Organização Mundial de Saúde e nas declarações da STOP-TB-INICIATIVE. Isso porque a estratégia DOTS exerce função profilática, dado que o tratamento diretamente supervisionado, negativa rapidamente os pacientes bacilíferos, cortando o elo da transmissão da doença, impede o aparecimento da resistência, atinge altos índices de cura, baixas taxas de abandono e poucas são as recaídas. Todavia, a estratégia DOTS está sendo difundida com muita dificuldade. Segundo a OMS, no ano de 2000, dos 211 países membros, apenas 127 (56,4%) aplicavam plenamente a estratégia DOTS, e somente 25% dos pacientes à recebiam. Nesse padrão, calcula-se que o DOTS só será totalmente adotado em 2003(223). Para a aplicação da estratégia DOTS, há necessidade da eficiência técnico-administrativa dos programas nacionais de controle da tuberculose, dentro da rede de saúde pública, o que está sendo denominado DOTS-PLUS( 224). Aqui entre nós, essa problemática foi analisada e avaliada nessa linha(225).
Contudo, a estratégia DOTS não soluciona os sérios problemas da tisiogênese com maior peso na epidemiologia da tuberculose, os quais decorrem da natureza do mecanismo imunitário: reinfecção exógena, reativação endógena, esta dependente da infecção latente. Dois bilhões de pessoas, um terço da humanidade, estão infectados com o Mycobacterium tuberculosis e com este convivem em simbiose. Urge uma vacina com poder de exaltar o processo imunitário e/ou de alterar sua qualidade, para tornar o organismo hospedeiro capaz de destruir o germe, estancando o estado de latência e com proteção suficiente para se defender contra as novas agressões externas.
Na atualidade, já são boas as perspectivas de que, com a engenharia genética, se construam vacinas sintéticas com eficácia para proteger positivamente contra os citados complexos problemas tisiogênicos. As expectativas na atualidade são ainda melhores graça a decifração do genoma do M. tuberculosis.
Decifração do genoma do mycobacterium tuberculosis
A história da tuberculose têm três ápices. A descoberta do agente causal em 1822, o advento da quimioterapia nos anos 40 do século 20 e o término da decifração do genoma do Mycobacterium tuberculosis em 1998.
Com a decifração do genoma do Mycobacterium tuberculosis abrem-se, afinal, novas perspectivas jamais imaginadas sobre o conhecimento dos intricados meandros da tisiogênese e de sua base imunitária. Do exposto nesta revisão, extrai-se a ilação de que não estão dominados com a necessária segurança, os mecanismos pelos quais células especializadas são mediadoras da imunidade da tuberculose. Nem se sabe com exatidão como os macrófagos destróem a micobactéria. As pesquisas (in vitro) e em "anima vili" empregando todos os elementos em jogo, foram de resultados precários permanecendo a incógnita do processo.
Com a decifração de genoma do M. tuberculosis, ingressamos na terceira mais importante fase da evolução da tuberculose.
Os responsáveis por essa histórica proeza da decifração do genoma do bacilo de Koch são, Cole ST e 41 associados, trabalho conjunto do Sanger Center, Instituto Pasteur, 6 outros centros e 100 laboratórios especializados(226).
A análise preliminar pode ser sintetizada como segue:
- O genoma do Mycobacterium tuberculosis contém 4000 genes, dos quais 3924 decodificados;
- São 4.411.529 pares de bases;
- O DNA contém 67% de guanina e citosina e 34% de adenina e timina;
- Proteínas conhecidas com precisão (40%), com algum conhecimento 44% e desconhecidos (16%);
- 140 reguladores da transcripção e mais 52 reguladores complementares explicam a elevada capacidade dos genes se adaptarem às interferências exteriores;
- Seqüências ricas em genes de biossíntese e de degradação de lipídios, revelam a complexidade da carapaça envolvente do germe, com ácido micólico, glicolípices, lipocarbohidratos. Mais de 250 genes fazem a biosssíntese das várias classes de lipídios;
- A inserção seqüencial IS 6110, as grandes seqüências de bases repetidas, os elementos ricos em guanina e citosina da família PGRS são utilíssimos para a fenotipagem das cepas pelas técnicas moleculares;
A perspectiva é de que se gastarão ainda alguns anos para o completo conhecimento dos elementos citados e para o seu aproveitamento prático para no controle da tuberculose de forma efetiva.
A decifração do genoma do Mycobacterium tuberculosis é a maior culminância de toda a história da pesquisa da tuberculose, depois de 116 anos de sua descoberta, e abre as portas de sua estrutura biofísica e sua significação. É a revolução que modificará a abordagem da tuberculose. Novos horizontes terapêuticos, com a identificação de enzimas e suas funções específicas; novas combinações técnicas biofísicas, novos modelos de atuação de drogas, inibidores de proteínas, abrindo novos ângulos de ataque à célula micobacteriana; conhecimento exato das bases moleculares da resistência do germe às drogas, os métodos para sua rápida detecção e para evitá-la; as drogas imunobiológicas no campo da imunoterapia com longa meia vida, possibilitando uma única administração ou com alguns intervalos longos. Outrossim, abrem-se novos e inesperados campos para a imunoprofilaxia, com vacinas poderosas desencadeando proteção absoluta contra o Mycobacterium tuberculosis. A decifração do genoma do M. tuberculosis, abre enorme leque de oportunidades inesperadas. Em síntese, enorme gama de oportunidades inesperadas.
Afinal, após mais de um século da descoberta do agente causal da tuberculose(227) e da obtenção da tuberculina com a qual Koch imaginou ter descoberto um remédio para a cura da tuberculose(228), que foi de resultados desastrosos, surgem esperançosas perspectivas para o seu efetivo combate, abrindo-se o caminho para a primavera de sua erradicação.
Referências bibliográficas
1. Calmette. L'infection bacillaire et la tuberculose chez I' homme et les animau. 4nd ed. Masson et Cie; 1936.
2. Canetti G, Diehl K, Good, et al. Advences in tuberculosis research. Vol. 8. New York: S Karger, Basel; 1945.
3. Rich AR. The pathogenesis of tuberculosis. 2nd ed. USA: Charles Thomas Ilinois; 1951.
4. Lopes JS, Rosember J. Estado atual, rumos e perspectivas da terapêutica biológica da tuberculose. Rev Paulista Tisiol 1937; 3-5.
5. Rosemberg J. Contribuição ao estudo da tisiogenese. Rio de Janeiro: Ass. Paulista de Medicina, Ed J do Brasil; 1941. [Premio Clemente Ferreira]
6. Madelaine C. Etudes sur la tuberculose. Paris; 1914. [Tese]
7. Lurie MB. Resistence to tuberculosis, experimental studies in native and acquired defensive mechanisms. USA: Harward Univ Press; 1964.
8. Yomans GP. Acquired immunity in tuberculosis J Chronic Dis 1957; 6: 606.
9. Seibert FB. A theory of immunity in tuberculosis. Perspect Biol Med 1960; 3:262.
10. Suter E. Multiplication of tubercle bacilli within mononuclear phagocytes in tissue cultures derived from normal animals and animals vaccinated with BCG. J Exp Med 1953; 97:235.
11. Mans GPY. Tuberculosis. Phliladelphia: W B Saunders; 1979.
12. Crowlw AJ, May MH. Preliminary demonstration of human tuberculoim munity in vitro. Infect Immun 1981; 31: 453.
13. Crowle AJ, Douvas GS, May MH. Naturaleza celular y molecular de la imunidad contra la tuberculosis en el hombre. Bol Union Int Tuber 1982; 58:75.
14. Organização Mundial de Saúde. Imunidad celular y resistencia a las infecciones. Informe Técnico 519: 1973.
15. Mackanes GB. Delayed hypersensitivity and the mecanism of cellular resistance to infection. Prog Immun 1971; 413.
16. Collins FM, Auclair LK. Effect of thymosin treatment on antituberlous immunity in immunosupressed mice. J Reticuloend Soc 1979; 26: 143.
17. Rook GAW. Importancia para la Union Internacional contra la Tuberculosis de ciertos progressos recientes em nuestra comprension de la inmunidad antimicrobiana mediada por celulas. Bol Union Int Tuber 1982; 58:62.
18. Zlotnik A, Yoshie. Chemocines: a new classification. Review and their role im immunity. Immun 2000; 12:121.
19. Schulger NW, Rom WN. The host immune response to tuberculosis. Am J Respir Crit Care Med 1998; 154: 679.
20. Boom WH. The role of T-ell subjects in Mycobacterium tuberculosis infection. Infect Agents Dis 1996; 5: 73.
21. Murray PJ. Defining The requirements for immunologicas control of mycobacterial infections. Trends Mycrobiology 1999; 7: 366.
22. Berman JSRL, Blumental H, Kornfeld JA et al. Chemotactic activity of mycobacterial lipoarabinomanan for human blood T lymphocites in vitro. J Immunol 1996; 156:3828.
23. Kurashima K, Mukaida N, Fusimura M, et al. Elevated chemocine levels in bronchoalveolar lavage fluid of tuberculosis patients. Am J Respir Crit Care Med 1997; 155: 1474.
24. Delespesse GCE, Demeure LP, Yangy ,et al. Maturation of naive human CD4 + T Linfocytes in to Th1, Th2 effectors. Int Arch Allergy Immunol 1997; 113; 157.
25. Stenger SRJ, Mazzaccaro K, Uyenura. Differential effect of cytolitic T cell subsets on intracellular infection. Science 1997; 276: 1684.
26. Byrd TF. Tumor necrosis factor alfa promotes grouwth of virulent Mycobacterium tuberculosis in human monocytes: iron- mediated growth suppression is correlated with decreased release of TNFg from iron- treated infected monocites. J Clin Invest 1997; 99:2518.
27. Kindler VAP, Sappino GE, Grau PF, et al. The inducing role of tumor necrosis factor in the development of bacterecidal granulomes during BCG infection. Cell 1989; 56: 731.
28. Flesh L, Kaufmann SH. Mycobacterium growth inibition by interferon-gama- activated bone marrow macrophages and differencial susceptisility among strain of Mycobacterium tuberculosis. J Immunol 1987; 138:4408.
29. Cooper AMDK, Dalton TA, Stewart JP, et al. Disseminated Tuberculosis in interferon gamma gene- disrupted mice. J Exp Med 1993; 178:2243.
30. Sodhi AJ, Gong C, Silva D, et al. Clinical correlated of interferon gama production with tuberculosis. Clin Infect Dis 1997; 25: 617.
31. Jaffe HAR, Buhal A, Mastrangeli KJ, et al. Organ Specific Cytokine therapy local activation of monuclear phogocytes by delivery of on aerosol of recombinant interferon gama to the human lung J Clin Invest 1991; 88: 297.
32. Holand SME, Eisenstein DB, Kuhns ML, et al. Tratment of refractor, disseminadated mycobacterial infection with interferon gamma. N Eng J Med 1994; 330:1348.
33. Condos RWN, Rom WN, Schuger W, et al. Treatment of multigrog resistent pulmonary tuberculosis with, interferon gamma via aerosol. Lancet 1997; 349:1513.
34. Dalton DK, Pitts-Meek S, et al. Multiple defects of immune cell function in mice with disrupted interferon-y genes. Science 1993; 259:1739-42.
35. Murray PJ, Yong RA, Daley QQ. Blood 1998; 91:2914.
36. Jouanguy E, Altare F, Lamhamedi S, et al. Interferon - y- recpetor deficieney in an infant whit fatal bacille Calmette Guerin infection. N Eng J Med 1996;335:1956.
37. Newport MJ, Huxley C, Huston S et al. A mutation in the interferon-y-receptor gene and susceptibility, to mycobacterial infection. N. Eng.Med.1996;335:1941.
38. Kaufmann SH. Gama/delta and other unconventional T'lymphocytes:what do they do? Proc Natl Acad Sci 1999; 93:2272.
39. Barnes PF, Grisso JS, Abrams H, et al. Gama Delta T linphocytes in human tuberculosis. J Infect Dis 1992; 165:506.
40. Ueta CI, Tsuyuguchi H, Kawasumi H, et al. Increase of gama/delta T cells in hospital workers who are in close contact whit tuberculous patients. Infect Immun 1994; 62:5434.
41. Label CHJ, Hess J, Daugelat P, et al. Contribuition of alpha/beta and gama/delta T lymphocytes to immunity against Mycobacterium bovis bacellus Calmette Guerin: studies with cell rectors- deficient mutant mice. Eur J Immunol 1995; 25:838.
42. Schlesinger L, Hull S, Kaufmant, et al. Binding of the terminal mannosyl unit of lipoarabino manan from a virulent strain of Mycobacterium tuberculosis to human macrophages. J Immunol 1994;152:4070.
43. Lalvani A, Pathan A, Durkan H, et al. Enhanned contact tracing spatial tracking of M. tuberculosis infection by enumeration of antigen-specific T cells, Lancet 2001; 357:2017.
44. Centers for Disease Control and Prevention-CDC. Prevention and tuberculosis among patiens infected with human immunodeficiency virus: principles of therepy and revised recommendations. Recommendations and reports. MMWR 1998; 47(RR-20):1-58.
45. Narita M, Ashkin D, Hollander ES, et al. Paradoxal worsening of tuberculosis following antiretroviral therapy in patients with AIDS. Am J Respir Crit Care Med 1998; 158:157.
46. Rich AR. Studies on the dissociation of hypersensitivity from immunity. Ver Immunol 1937; 3:25.
47. Yuumans GP. Tuberculosis. Development of delayed tuberculin hipersensitivity in tuberculosis. Philadelphia: Senders; 1979.
48. Mackaness GB. Artificial cellular immunity against tubercle bacile. Am Rev Tuber 1954; 69:690.
49. Metaxas MN, Metaxas M. Studies on the passive transfer of tuberculosis sensitivity. Immunopathology. First International Symposium Basel, 1958. Basel: Graber, Miescher; 1959.
50. Sever JL. Passive transfer of resistence to tuberculosis through use of monocites. Proc Soc Exp Biol Med 1960; 103: 326.
51. Nowakowski MSP, Cham P, Steiners, et al. Different distribuition of lung and blood subsets in pediatric AIDS or tuberculosis. Ann Clin Lab Sci 1992; 22: 377.
52. Hoheisel GBL, Tabak H, Teschler F, et al. Bronchoaveolar lavage cytology, and immunocitology in pulmonary tuberculosis. Am J Respir Crit Care Med 1994; 149: 460.
53. Yu CT, Wang TJ, Huang HC, et al. Relation of broncoalveolar lavage T Lymphocyte subpopulation to rate of regression of active tuberculosis. Thorax 1995; 50: 869.
54. Schlesinger LS. Entry of Mycobacterium tuberculosis into mononuclear phagocytes. Curr Top Microbiol Immunol 1996; 21: 71.
55. Jackett PS, Aandrew PW, Aber VR, et al. Les macrophages alveolaires du cobaye tuent probablement M. Tuberculosis, H37Rv et H37Ra in vivo en produisant du peroxide d'hydrogene. Bull Union Int Tuberc 1983; 58: 223.
56. Walker L, Lowrie DB. Macrophages peritoneaux de souris tuant Mycobacterium microti par production de peroxyde d'hidrogene. Bull Union Int Tuberc 1983; 58: 229.
57. Gordon AH, Hart PD, Young R, et al. Nature 1980; 286:79.
58. Cham J, Xing Y, Magliozzo RS, et al. Killing of virulent Mycobacterium tuberculosis by reactive nitrogen intermediates produced by activated murine macrophages. J Exp Med 1992; 175: 1111.
59. Mac Micking JDR, Nortj R, La Course JS, et al. Identification of nitric. oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci USA 1997; 94: 5243.
60. Hoheisel GL, Zheng H, Tescheler L, et al. Incrised soluble CD14 levels in BAL fluid in pulmonary tuberculosis. Chest 1995; 108: 1614.
61. O'Brien L, Roberts B, Aandrew PW, et al. - In vitro interation of macrophages and mecanism of anti - mycobacterial activity. Curr Top Microbiol Immunol 1996; 215: 97.
62. Rhoades ER, Cooper AM, Orme JM. Chemokine response in mice infected with Mycobacterium tuberculosis. Infect Immunty 1995; 63: 3871.
63. Nicholson S, Bonecine MGA, Silva RL, et al. Inducible nitric oxide synthase in pulmonar alveolar macrophages from patients with tuberculosis. J Exp Med 1996;183: 2293.
64. Adams DO. The granulomatous inflomatory response. A review. Am Pathol 1976; 84: 161.
65. Molleoy A, Kaplan G. Apoptosis but not necrosis, of infected monocytes is coupled with killing intracellular bacilus Calmette Guerin. J Exp Med 1994; 180: 1499.
66. Reed JC. Mini- review: cellular mechanism of diseases series. Bcl- 2 and the regulation of programmed cell death. J Cell Biol 1994; 124: 1.
67. Korsmeyer SJ. Regulators of cell death. Tig 1995; 11: 101.
68. Cree IA, Nurbhai S, Milne G, et al. Cell death in granulomata: the role of apoptosis. J Clin Pathol 1987; 40: 1319.
69. Klinger K, Kam-Meng W, Brandli O, et al. Effects of mycobacteria on regulatin of apoptosis in mononuclear phagocytes. Infect Immun 1997; 65: 5272.
70. Vaux DL. Toward an understanding of the molecular mechanisms of physiological cell death. Proc Natl Acad Sci USA 1993; 90: 786.
71. Raviglione ME, Snider DE, KochiI. Global epidemiology of tuberculosis. Morbidity and mortality of a world wide epidemic. JAMA 1995; 273: 220.
72. Dolin PJ, Raviglione MC, Kochl. Global tuberculosis incidence and mortality during 1990-2000. Bull Word Health Organ 1994; 731: 213.
73. Orme IM. The latent tuberculosis bacillus ( I'll let you know if ever meet one ). Int J Tuberc Lung Dis 2001; 5: 589.
74. Yuan Y, Crane DD, Simpson RM, et al. The 16kDa alfa cristalin ( Acr) protein of Mycobacterium tuberculosis is required for growth in macrophages. Proc Natl Acad Sci USA 1998; 95: 9578.
75. Waine LG. Dormance of Mycobacterium tuberculosis and latency of disease. Eur J Clin Microbiol Infect Dis 1994; 13: 905.
76. Waine LG, Lin KY. Glyoxilate metibolism and adaptation of Mycobacterium tuberculosis to survival under anaerobic condition. Infect Immun 1982; 34: 104.
77. Waine LG, Hayes LG. An in vitro model for sequencial study of shiftdown of Mycobacterium tuberculosis through two stages of non replicating persistence. Infect Immun 1996; 64: 2062.
77A- Grange JM. The mystery of the "mycobacterial persistor". Tuber Lung Dis 1992; 73:299.
78. Waine LG, Hayes LG. Nitrate reduction as a maker for hipoxic shiftdown of Mycobacterium tubeculosis. Tuberc Lung Dis 1998; 79: 127.
78A- Rist N. Tese da Faculdade de Medicna da Universidade de Paris. 1938.
78B- Saens A, Canetti G. Presse Med 1939; 27: 5.
78C- Rosemberg J. O poder patogênico e alergisante do bacilo de Koch morto e o atual conceito de virulencia do agente causal da tuberculose. Rev Bras Tuberc 1941; 7: 331.
79. Paraf J. L'immunité au cour de la tuberculose. Étude experimental e et clinique. Paris: Masson Cie; 1936.
80. Gardner L U. The history of the R1 strain of tuberculosis bacillus. Am Rev Tuberc 1932; 25: 577.
81. Steenken W, Gardner LU. History of H37 strain of tubercle bacillus. Am Rev Tuber 1946.
82. Schesinger LS. Macrophage phagocitosis of virulent but not attenuated strain of Mycobacterium tuberculosis is mediated by manose receptors in addition to complement receptors. J Immunol 1993; 150: 2920.
82A- Prabhakar R, Venkatraman P, Vallishayee RS, et al. Virulence for guinea pigs of tubercle bacilli isolated from the sputum of participant in the BCG Trial. Cingleput. District, South India. Tubercle 1984; 68: 3.
82B- Thierry D, Brisson-Noel A, Frebault VV, et al. Caracterization of a Mycobacterium tuberculosis insertion sequence, IS 6110, and its application in diagnosis. J Clin Microbiol 1990; 28: 2668.
82C- Goial M, Saunders NA, Yan Embdem JDA, et al. Differentiation of Mycobacterium tuberculosis isolates by spoligotyping and IS6IIO restriction fragment length polymorphism: J Clin Microbiol 1997; 35: 647.
82D- Van Embdem JDR, Gorkom T, Kremer K, et al. Genetic variation and evolutionary origin of the Direct Repeat locus of Mycobacterium tuberculosis complex bacteria. J Bacteriol 2000; 182: 2393.
83. Fine PEM. Immunities in and to tuberculosis: implications for pathogenesis and vaccination, in tuberculosis, back to the future. New York: J Porter; 2000.
84. Fomurong NG, Dale JW, Osborn TWE, et al. Use of gene probes based on the insertion sequence IS 986 to differenciate between BCG vaccine strains. J Apl Bacteriol 1992; 72: 126.
85. Behr MA, Wilson MA, Gill WP, et al. Comparative genomic of BCG vaccines by whole- genome DNA. Microarray. Science 1999; 284: 1520.
86. Aboud-Zel C, Smith J, Grange JM, et al. Subdivision of daughter straim of bacille Calmette- Guerin (BCG) according to secreted protein patterns. J Gen Microbiol 1986; 132: 3047.
87. Aston CW, Rom WN, Talbot AT, et al. Differential killing of Mycobacterium bovis (BCG) by normal human neutrophils, monocytes and alveolar macrophages; a role for defensins? Am J Respir Crit Care Med 1995; 151: A245.
88. Molloy A, Roonlvorapomg PL, Kapzan G, et al. Apoptose but not necrosis, of infected monocytes is coupled with killing of intracellular bacillus Calmette- Guerin. J Exp Med 1994; 180: 1499.
89. Adams D. The structure of mononuclear phagocytes differentiating in vivo: I- Sequencial fine and histologic studies of the effect of bacillus Calmette-Guerin (BCG). Am J Pathol 1974; 76: 17.
90. Dubos RE. Propecties and structures of tubercle bacilli concerned with their pathogenicity. Symp Soc Gen Microbiol 1955; 5:103.
91. Molloy A, Meyn PA, Smidt KD, et al. Recognition and destruction of bacillus Calmette- Guerin infected human monocytes. J Exp Med 1993; 177: 1691.
92. Kumararatne DS, Pithieas, Drysdale P, et al. Specific lysis of mycobacterial antigenbearing macrophages by class 2 MHC- restrited polyclonal T cell lines in healthy donors or patients whit tuberculosis. Clin Exp Immunol 1990; 80: 314.
93. Valway SE, Sanchez MP, Shinnick TF, et al. An outbreack involving extensive transmission of a virulent strain of Mycobacterium tuberculosis. N Eng J Med 1998; 338: 693.
94. Manca C, Tsenoval L, Barry CE, et al. Mycobacterium tuberculosis CDC 1551 induces a more vigorous host response in vivo and in vitro, but is not more virulent than others clinical isolates. J Immunol 1999; 162: 6740.
95. Barnes P, Yang Z, Preston-Martin S, et al. Patterns of tuberculosis transmission in central Los Angeles. JAMA 1997; 278: 1159.
96. Zhang M, Gong J, Yang Z, et al. Enhanced capacity of a widespread strain of Mycobacterium tuberculosis to grow in human macrophages. J Inf Dis 1999; 179: 1213.
97. Collins D, Kawakami RP, Lisle G, et al. Mutation of principal factor causes loss of virulence in a strain of the Mycobacterium tuberculosis complex. Proc Natl Acad Sci USA 1995; 92: 8036.
97A- Middlebrook G, Cohn MN. Some observation on the pathogenecity of isoniazid- resistent variants of tubercle bacilli. Science 1953; 118: 297.
97B- Long ER. The chemestry and chemotherapy of tuberculosis. Baltimore: W. Wilkins Co ; 1958.
98. Rieder HL. Bases épidemiologiques de la lutte antituberculeuse. Union Intenationale Contre la Tuberculeuse et les maladies Respiratoires. vol. 1; 1999.
98A- Warburton ARE, Jenkins PA, Waight PA, et al. Drug resistence inicial isolates of Mycobacterium tuberculosis in England and Wales. Commun Dis Resp 1993; 3 R 175.
98B- Frieden TR, Sterling T, Mendes AP, et al. The emergence of drug- resistant-tuberculosis in New York City. N Eng J Med 1993; 318: 521.
98C- Moss AR, Alland D, Telza KE, et al. A city-wide outbreak of a multiple drug-resistant strain of Mycobacterium tuberculosis in New York City. Int J Tuberc Lung Dis 1997; 1: 115.
98D- Mitchison DA, Nunn AJ, Influence of initial drug resistence on the response to short-course chemotherapy of pulmonary tuberculosis Am Rev Respir Dis 1986; 133: 423.
98E- Melo FAF, Neto JI, Seiscento M, et al. Tuberculose multirresistente. J Pneumol. 1993; 19; 73.
99. Comstock CW. Tuberculosis in Twins Am Rev Respir Dis 1978; 117: 621.
100. Stead WW, Senner JW, Reddick WT, et al. Racial differences in susceptibility to infection by Mycobacterium tuberculosis. N Eng J Med 1990; 322: 422.
101. Stead WW. Genetic and resistence to tuberculosis. Ann Int Med 1992; 116: 938.
102. Stead WW, Lofgren JP, Senner JW, et al. Relative susceptibility of black American to tuberculosis. Am J Epidemiol 1994; 139: 531.
103. Mays EE . Pulmonary diseases. In: Textbook of black- related diseases. New York: Willians RA; 1975.
104. Crowle AJ, Elkins N. Relative permissiveness of macrofages from black and white people for virulent tuberculosis bacilli. Infect Immun 1990; 58: 632.
105. Rook GAW. The role of vitamin D in tuberculosis. Am Rev Respir Dis 1988; 138: 770.
106. Bifani PJ, Plikaytis BB, Kapur V, et al. Origin and interstate spread of a New YorK City multidrog-resistant Mycobacterium tuberculosis. JAMA 1996; 275:425.
107. Mehra NK, Bovornkitti S. HLA and tuberculosis-a reapraisal. J All Immunol 1986; 4: 149.
108. Xingpei X, Senbin L, Chaoyng W, et al. Study on the association of HLA with pulmonary tuberculosis. Immunol Invest 1986; 15: 327.
109. Arif AL, Goldstein RA, Afronti LF, et al. HLA B15 and tuberculosis in North American black population. Am Respir Dis 1979; 120: 1257.
110. Selby R, Barnard JM, Buehler SK. Tuberculosis associated with HLA- B8, Bf S in a Newfound land Community study. Tissue Antigens 1978; 11: 403.
111. Singh SPN, Mehra NK, Dingley HB et al. HLA- A- B- C and DR antigen profile in pulmonary tuberculosis in North India. Tissue Antigens 1983; 21: 380.
112. Khomenko AG, Litvinov Vi, Cukanova VP, et al. Tuberculosis in patients with varius HLA phenotypes. Tubercle 1990; 71: 187.
113. Hawkins BR, Higgins DA, Chan SL, et al. HLA typing in the Hong Kong Chest Service/ British Medical Research Cousil study of factors associated with breakdown of active tuberculosis of inactive pulmonary lesions. Am Respir Dis 1988; 130: 1616.
114. Belalamy R, Ruwend C, Corrach T, et al. Variation in the NRAMP1 - Gene and Susceptibility to tuberculosis in west africans. N Eng J Med 1998;338:640.
115. Vidal S, Tremblay ML, GovoniI G, et al. The Ity/Lsh/Bcg locus. Natural resistence to infection with intracellular parasites is abrogated by ddisruption of the NRAMP1. J Exp Med 1995;182:155.
116. Vidal S, Pinner E, Lepage P, et al. Natural resintece to intracelular infections. Nramp1 encodes a membrane phosphoglycoprotein absent in macrophages from susceptible Nramp1 mouse strains. J immunol 1996;157:3559.
116A- Vidal S, Malo D, Vogan K, et al. Natural resistence to infection with intracelular parasites: isolation of a candidate for Bcg-r. Cell 1993;73:469.
117. Liu J, Fujiwara M, Buu N, et al. Identification of polymorphisms and sequence variants in the human homologue of the mouse natural resistence - associated macrophage protein gene. Am J Human Genetic 1995; 56:845.
118. North RJ, Medina E. Who important is Nramp1 in tuberculosis? Trends Microbiol 1998; 6:441.
119. Bellamy R, Ruwende C, Corbah T, et al. Variations in the NRAMP1gene and susceptibilty to tuberculosis in west Africa. N Eng J Med 1998;338:640.
120. Newport, Levin M, Shaw MA, et al. Evidence for exclusion of a mutation in rmap1 as the cause of familial disseminated atipical mycobacterial infection in Maltese kindred J Med Gent 1995; 32;904.
121. Harboe M, Oetinger T, Wiker HG, et al. Evidence for ocurrence of the ESAT-6 protein in mycobacterium tuberculosis and virulent mycobacterium bovis and for its abscence in mycobacterium bovis BCG. Immunity 1996;64:16.
122. Bretscher PA. A strategy to improve the efficacy of vacination against tuberculosis and leprosy, Immunol Today 1992; 13:342.
123. Kindler V, Sappino AP, Grau GE, et al. The inducing role of tumor necrosis factor in the development of bacterecidal granulomas during BCG infection. Cell 1989; 56:731.
124. Jackett PS, Bothamely GH, Batra HV et al. Specifivity of antibodies to immunodominant mycobacterial antigens in pulmonary tuberculosis. J Clin Microbiol 1988; 26:2318.
125. Kaufmann SHE. CD8.T - lymphocytes in intracellullar microbial infections. Immunol Today, 1988; 9:168.
126. Kaufmann SHE. Immunity to intracellular bacteria. Annu Rev Immunol 1993; 11:129.
127. Chan JY, Xing Y, Maglioso RS, et al. Killing of virulent M. tuberculosis by reative Nitrogen intermediate produced by activated murine macrophages. J Exp Med 1992; 175:1111.
128. Scanga CA, Mohan VP, Joseph H, et al. Reativation of latent tuberculosis variations of the Cornell murine model. Infect immun 1999; 67:4531.
129. Stead WW. Paphogenesis of the first episode of chonic pulmonary tuberculosis in mam; infection or exogenous reinfection? Am Rev Respira Dis 1967; 95:729.
130. Flyn JL, Scanga CA, Tanaka KE, et al. Effets of aminoguanidine of latent murine tuberculosis. J Immunol 1998; 160:1796.
131. Orme JM. A mouse model of the reerudescence of latent tuberculosis in the elderly. Am Rev Respir Dis 1998; 137:716.
132. Stead WW. Genetic and resisttance to tuberculosis. Ann Intern Med 1992; 116:937.
133. Clak GA, Kelley MA, Grange JM, et al. The evolution of Mycobacterial disease in human populations. Current Antropol 1987; 28:45.
134. Bates JH, Stead WW. The history of tuberculosis as a global epidemic. Med Clin North Amer 1993; 77:1205.
135. Sjogren I, Sutherland I. Studies of tuberculosis in man relation to infection in catlle. Tubercle 1974; 56:113.
136. Davies RPO, Tocque K, Bellis MH, et al. Historical declines in tuberculosis in England and Wales: improving social conditions or natural selection. Int J Tuberc Lung Dis 1999; 3:1051.
137. Webb GB. Tuberculosis. New York: C. Medicine; 1936.
138. Livingstone D. Missionary travels and researches in south Africa. Ward Lock; 1857.
139. Hirsch A. Hand book of geographical and historical pathology. Vol. III New Sydenham Society; 1886.
140. Cummins SL. Primitive tuberculosis. J Bale Med Pub 1939; 14:22.
141. McCaulay D. Budd W. On the nature and propagation of phtisis. Lancet 1967; 2:45.
142. Tuberculosis in the Egyptian army. Brit J Tuber 1908; 2:35.
143. Wilkinson E. Notes on the prevalence of tuberculosis in India Proc Royal Soc Med 1914; 8:195.
144. Brown P, Cathal F, Gajdusek DC. Mycobacteria sensitivity patterns among remote population groups in Papua, New Guinea and in the herbides, Solomon and Caroline Island. Am J Trop Med Hyg 1981; 30:1085.
145. Fergunson RG. Studies in tuberculosis. Toronto: Univ Tor Press; 1955.
146. Neves JS. A outra historia da Companhia de Jesus. Vol.1. Vitória, Espírito Santo; 1984.
147. Osler W. The principles and practice of medecine. Vol. 1. Ed. Appleton; 1892.
148. Karasch MC. A vida dos escravos no Rio de Janeiro. 1808-1850. Vol. 1. Comp. das Letras: 2000.
149. Borel A. Pneumonie et Tuberculose ches les troupes noire. Ann Inst Pasteur 1920; 34:105.
150. Kiple KF. Another dimension to the black diaspora. Diet, disease and racism. Vol. 1. Cambridge: Univ.Press; 1981.
151. McCartthy FP. The influence of race in the prevalence of tuberculosis. Med Surg J 1912; 166:207.
152. O'Brien SJ. Ghetto legacy. Mol Evotution 1991; 1:209.
153. Osborn TW. Changes in BCG straim. Tubercle 1983; 64:1.
154. Comstock GW. Identification of an effective vaccine against tuberculosis. Am Rev Respir Dis 1988; 138:479.
155. Who Expert Committee on Tuberculosis. Eigth Report. Technical Report Series, n.290. 1964.
156. Hermans PWM, Soolingen D, Dale JW, et al. Insertion element IS 986 from Mycobacterium tuberculosis. A useful tool diagnosis and epidemiology of tuberculosis, J Clin Microbiol 1990; 28:2051.
157. Bloom BR, Fine PEM. The BCG experience: implications for future vaccines against tuberculosis. Pathogenesis, protection and control. USA: B.R.Bloom/ Am Soc Microbiol; 1994.
158. Orme IM. Prospects for new vaccines against tuberculosis. Trends Microbiol 1995; 3:401.
159. Orme IM. Progress in the development of new vaccines against tuberculosis. Int J Tuberc Lung Dis 1997; 95:100.
160. Poulet S, Cole ST. Characterization of the polymorphic GC rich repetitive sequence (PGRS) present in Mycobacterium tuberculosis. Arch Microbiol 1995; 163:87.
161. Behr MA, Wilson MA, Gill WP, et al. Comparative genome of BCG vaccine by whole- DNA microarray. Science 1999; 284:1520.
162. Mahiras GG, Sabo PJ, Hickei MJ, et al. Molecular annalysis of genetic difference between Mycobacterium bovis BCG and virulent Mycobacterium bovis. J Bacteriol 1996; 178:1274.
163. Palmer CE, Long MW. Effects of infection with atypical mycobacteria on BCG vaccination and tuberculosis. Am Rev Respir Dis 1996; 94:553.
164. Constock GW, Livesay VT, Woolper SF, et al. Evaluation of BCG vaccination among Porto Rico children, Am J Pub Health 1974; 64:283.
165. Dam HC, Hitze KL. Vaccunation com BCG de reciem nascidos y latentes. Who U IT tuberc ( WHO/TB/76.104).
166. Clemens JD, Chuang JHJ, Feinstein N. The BCG controversy. a methodological and statiscal reapraisal. JAMA 1983; 249:2362.
167. Ponnighaus JM, Fine PEM, Sterne JAC, et al. Efficacy of BCG vaccine against leprosy and tuberculosis in northern Malawi. Lancet 1992;339:636.
168. Karonga Prevention Trial Group. Randomised controlled trial of single BCG repeated BCG or combined BCG and killed Mycobacterium leprae vaccine for prevention of leprosy and tuberculosis in Malawi. Lancet 1996; 348:17.
169. Romanus V. Tuberculosis in bacillus Calmette Guerin immunized en children sweden. A ten year evaluation following the cessation of general vaccination in new born in 1945. Bull Union Tuberc Mal Respir 1990; 65:31.
170. Hermans PWM, Van Solongen D, Bik EM, et al. Insertion element IS 987 from Mycobacterium bovis BCG is located in a hot- spot integration region for insertion elements in Mycobacterium tuberculosis complex straims. Infect Immun 1991; 59:2695.
171. Lugosi L. Resultado de analises de la eficacia de la vacinacion sistematica com BCG de 1950 a 1983 em el control de la tuberculosis. Bull Int Union Tuberc Mal Respir 1987 ;62:10.
172. 2o Informe Técnico Sobre Vacinação e Revacinação BCG. CNPS. Brasília: 1994.
173. Rodrigues LA, Diwan VK, Waeller JB. Protective effect against tuberculosis meningites and millary tuberculosis. A meta-analysis of the published literrature. Int J Epidemiol 1993;22:1154.
174. Colditz GA, Brewer TF, Berrey CS, et al. Efficacy of BCG vaccine in the prevention of tuberculosis. Meta analysis of the published literature. JAMA 1994; 271:699.
175. Cohn DL, O'Brien RJ. The use of restriction fragment length polymorhism (RFLP) analysis for epidemiological studies of tuberculosis in developing countries. Int J Tuberc Lung Dis 1998;2:16.
176. Solingen VD, Qianl HAAS, et al. Predominant genotipe of Mycobacterium tuberculosis in countries of Eest Asia J Clin Microbiol 1995; 33:3234.
177. Cheng SH, Walker KB, Lowrie DB, et al. Monocytes antibacterial activity before and after Mycobacterium bovis BCG vaccination in Chingleput, India, and London, United Kingdom.Infect Immun 1993; 61:4501.
178. Parronchi P, Macchia D, Piccin P, et al. Allergen and bacterial antigen-specific T Cell clones established from atopic donnors show indifferent profile of cytokine production. Proc Natl Acad Sci USA 1991; 88:45.
179. Salgame P, Abrams J, Clayberger C, et al. Differing lymphokine profiles of functional subsets of human CD4 and CD8 T cell clones. Science 1991: 254:279.
180. McDonough KA, Kress Y, Bloom BR.. Pathogenesis of tuberculosis interaction of Mycobacterium tuberculosis with macrophages. Infect Immun 1993; 27:63.
181. Rodrigues LC, Gill N, Smith PG, et al. BCG vaccination in the first year of life protects children of Indian subcontinent ethnie origin against tuberculosis in England. J Epidemiol Comm Health 1991;45:78.
182. Crowle AJ. Immunization against tuberculosis what kind of vaccine? Infect Immun 1988; 56:69.
183. Rook G. Progress in the immulogy of the micobacteriosis. Clin Exp Immunol 1986; 132:3047.
184. Fine PEM, Rodrigues LC. Modern vaccines. Mycobacterial diseases. Lancet 1990; 335: 1016.
185. Advisoty Counsil for the Elimination of Tuberculosis (ACET). Development of new vaccines for tuberculosis. MMWR 1998; 47: 1.
186. Bloom BR, Jacob WR. New strategies for Leprosy and Tuberculosis and for development of bacillus Callmette-Guerin into a multivaccine vehicle. New York: Acad Science, Dep. Microbiology and Immunology; A. Einstein College Medicine; 1990.
187. Aldovani A, Young RA. Humoral and cell mediated immune responses to live recombinant BCG - HIV vaccines. Nature 1991; 551: 479.
188. Orme IM. Prospects for new vaccines against tuberculosis. Trends Microbiol 1995; 3: 401.
189. Cirillo JD, Stover C, Bloom BR, et al. Bacterial vaccine vectors and Callmette-Guerin. Clin Infect Dis 1995; 20: 1001.
190. Van Vooren JP, Drowart A, Bruyn J, et al. Humoral responses against the 85 A and 85 B antigens of Mycobacterium bovis BCG in patients with leprosy and tuberculosis . J Clin Microbiol 1992; 30: 1608.
191. Huygen K, Content J, Denis O, et al. Immunogenicity and protective efficacy of a tuberculosis DNA vaccines . Nat Med 1996; 2: 893.
192. Fine PEM. Variations in protection by BCG: Implications of and for heterologous immunity. Lancet 1995; 346: 1339.
193. Harboe MH, Oettinger T, Wiker HG, et al. Evidence for ocurrence of the ESAT-6 protein in Mycobacterium bovis and for its absence in Mycobacterium bovis BCG. Infect Immun 1996; 64: 16.
194. Arend SM, Ottenhoff M, Andersen P, et al. Uncommon presentations of tuberculosis : The potencial value of a novel diagnostic assay based on the Mycobacterium tuberculosis - specific antigens ESAT-6 and CFP- 10. Int J Tuberc Lung Dis 2001; 5: 680.
195. Lalvani A, Pathan AA, Durkan H, et al. Enhanced contact tracing and spatial tracking of Mycobacterium tuberculosis infection by enumeration of antigen- specific T cells. Lancet 2001; 357: 2017.
196. Launois P, De Leys R, Niang M, et al. T- cell epitopes mapping of the mafor secreted mycobacterial antigen 85 in tuberculosis and leprosy. Infect Immun 1994; 62: 3679.
197. Stover CK, Bansal GP, Hanson MS, et al. Prospective immunity elicit by recombinant bacille Callmette- Guerin (BCG) expressing on their surface protein A (OspA) lipoprotein: New use, of BCG for recombinant vaccines. Nature 1991; 351: 456.
198. Lugosi L, Jacob's WR, Bloon BR. Genetic transformation of BCG. Tubercle 1989; 70: 159.
199. Orme IM. Progrers in the development of new vaccines against Tuberculosis. Int J Tuberc Lung Dis 1997; 1: 95.
200. Collins FA. Anitituberculosis immunity: new solutions to an old problem. Rev Inf Dis 1991; 13: 940.
201. Hart PD, Sutherland I, Thomas J. The immunity conferred by effective BCG and vole bacilus vaccines, in ralation to the induced tuberculin sensitivity and to technical variations in the vaccines. Tubercle 1967; 48: 201.
202. Hart PD, Sutherland I. BCG and vole bacillus vaccines in the prevention of tuberculosis in adolescence and early adult life. Brit Med J 1977; 22: 293.
203. Brown CA, Brown IN, Swinburne S. The effect of oral Mycobacterium vaccae on subsequent responses to BCG sensitization. Tubercle 1985; 66: 251.
204. Onyebujoh P, Rook GAW. Mycobacterium vaccae immutherapy. Lancet 1991; 338: 1554.
205. Stanford JL, Rook GAW, Bahr GM, et al. Mycobacterium vaccae in immunoprophylaxis and immunotherapy of leprosi and tuberculosis . Vaccines 1990; 8: 525.
206. Phillip WJ, Poulet S, Eiglmeier K, et al. An integrated map of the genome of the tubercle bacillus, Mycobacterium leprae. Proc Natl Acad Sci USA 1996; 93: 3122.
207. Yong DB, Cole ST . Leprosy, tuberculosis and new genetic. J Bacteriol 1993; 175: 1.
208. Fine PEM. BCG vaccination agains tuberculosis and leprosy. Brit Med J 1988; 44: 691.
209. Sharma P, Misra RS, Kar K, et al. Mycobacterium W vaccine, a useful adjuvant to multidrug therapy in multibacillary leprosy. Lepr Rev 2000; 71: 79.
210. Reddy PP, Amin AG, Khaswkar OS, et al. Molecular definition of unique species status of Mycobacterium W: a candidate leprosy vaccines strain. Int J Lepr 1994; 62: 229.
211. Winter N, Lagranderie J, Rauzier J, et al. Expression of heterologous genes in Mycobacterium bovis BCG induction of celular response against HIV-1 Nef protein. Gene 1991; 109: 47.
212. Bange FC, Brown AM, Jacob WR. Leucine auxotrophy restrict growth of Mycobacterium bovis BCG in macrophages. Infect Immun 1996; 64: 1794.
213. Huyguen K. DNA vaccines: application to tuberculosis. Int J Tuberc Lung Dis 1998; 2: 971.
214. Wolff JA, Malone RW, Williams P, et al.. Direct gene transfer into mouse muscle in vivo. Science 1990; 247:1465.
215. Andersen P. Effective vaccination of mice against Mycobacterium tuberculosis infection with a soluble mixture of secreted Mycobacterial proteins. Infect Immun 1994; 62: 2536.
216. Huygen K, Content J, Denis O, et al. Immunogenicity and protective efficacy of a tuberculosis DNA vaccine. Nat Med 1996; 2: 893.
217. Ulmer JB, Liu MA, Montgomery DL, et al. Expression and immugenity of Mycobacterium tuberculosis antigen 85 by DNA vaccination. Vaccine 1997; 15:792.
218. Tacson RE, Colston MJ, Ragno S, et al. Immunugenicity and protective efficacy of a tuberculosis DNA vaccine. Nat Med 1996; 2:893.
219. Bonato VLD, Lima WMF, Tacson RE, et al. Identification and characterization of protective T cells in hsp 65 DNA vaccinated and Mycobacterium tuberculosis infected mice. Infect Immunity 1998; 66:169.
220. Enserink M. Driving a stake into resurgent TB. Science 2001; 293:234.
221. Silva CP. Desenvolvimento de novas vacinas para a tuberculose. III Forum Estadual de Combate à tuberculose. João Pessoa, Paraíba: 19-22 nov. 2001.
222. Bishai W. Lipid Lunch, for persistent pathogen. Nature 2000; 406:683.
223. Global tuberculosis Control. Who report 2001. Geneve: World Health Organization; 2001.
224. Farmer PE, Kim JY. Community - bases aproaches to the control of multidrug-resistant tuberculosis. Introducing DOTS-PLUS. Br Med J 1998; 317:67.
225. Teixeira GM. DOTS - Uma estratégia renovada. Bol Pneum Sanit 1998; 6:3.
226. Cole ST, Brosch. R, Parkhill J, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998; 393:537.
227. Koch R. Die aectiologie der Tuberkulose. Berl Klin. Wochenschr 1822; 19:221.
228. Koch R. Weitere mitheilunger uber ein heilmittel gegen tuberkulose. Dtsch Med Wochenschr 1890; 16:1029.