









 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Curriculum ScienTI
Curriculum ScienTI Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntrodução
A Organização Mundial da Saúde (OMS) define anomalias congênitas (defeitos congênitos) como alterações estruturais ou funcionais no embrião ou feto, derivadas de fatores anteriores ao nascimento, possíveis de se identificar no acompanhamento pré-natal, no nascimento ou mais tarde, ao longo da vida.(1) No mundo, estima-se cerca de 3% dos nascidos vivos com alguma anomalia, a cada ano,(2),(3) sendo que pelo menos 3,3 milhões de crianças menores de 5 anos morrem anualmente, por causa de anomalias congênitas graves. Entre os afetados que sobrevivem, muitos apresentam incapacidades durante a vida.(4)
Um sistema de codificação e classificação abrangente e robusto é essencial para a vigilância de anomalias congênitas, porém não existe uma lista acordada que reúna todos esses agravos.(5) A Classificação Estatística Internacional de Doenças e Problemas Relacionados à Saúde (CID) constitui a codificação adotada internacionalmente, para propósitos epidemiológicos. Em sua décima revisão (CID-10), seu capítulo XVII (Malformações congênitas, deformidades e anomalias cromossômicas) faz referência exclusivamente a anomalias congênitas.(6) Apesar de esse capítulo ser amplamente utilizado como referência para vigilância, existem outros agravos que se enquadram no conceito de anomalias congênitas mas se encontram em outros capítulos da mesma CID-10. A despeito da atualização realizada em sua décima primeira edição, CID-11, instituições internacionais, como a International Clearinghouse for Birth Defects Surveillance and Research (ICBDSR) e a European Network of Population-Based Registries for the Epidemiological Surveillance of Congenital Anomalies (EUROCAT), apontam que a última versão não será suficiente para englobar todas as anomalias congênitas em capítulo único.(5)
Alguns programas internacionais de vigilância de anomalias congênitas incluem condições ausentes do capítulo XVII da CID-10, segundo seus objetivos e prioridades. No Brasil, os códigos do capítulo XVII da CID-10 são utilizados para registro dos casos de anomalias congênitas em sistemas de informações, a exemplo do Sistema de Informações sobre Nascidos Vivos (Sinasc). A proposição de uma lista utilizando-se de múltiplas fontes de informação visa permitir o estabelecimento de um conjunto ampliado de anomalias congênitas, de maneira a elencar aquelas prioritárias para monitoramento. Desta forma, será possível expandir o rol de códigos habilitados para inserção no sistema e, por conseguinte, aprimorar a vigilância de anomalias identificadas no nascimento.
Neste trabalho, objetivou-se propor uma lista expandida de anomalias congênitas para além do capítulo XVII da CID-10, visando a sua disponibilização para aplicação no âmbito da vigilância em saúde.
Construção da lista
Foram utilizadas as seguintes fontes de informação: CID-10, publicada em 2007 e vigente no Brasil; CID-11, lançada pela OMS em 2019, com o propósito de ser implementada a partir de 2022; anomalias congênitas monitoradas por modelos de vigilância selecionados, identificados a partir de uma revisão narrativa da literatura;(7) e um repositório/base de informações sobre doenças raras, o Orphanet.(8) Realizou-se a extração dos códigos das diferentes fontes em dezembro de 2019.
Foram incluídas condições que atendessem ao conceito de anomalias congênitas adotado pela OMS.(1) Embora não sejam anomalias congênitas por definição, as infecções congênitas foram consideradas, haja vista algumas apresentarem desfechos importantes no contexto da vigilância.(9) Foram excluídas condições sobre as quais não havia codificação correspondente na CID-10, especialmente para manter correspondência com o registro dos casos nos sistemas de informações do Ministério da Saúde.
Os códigos das anomalias congênitas do capítulo XVII foram captados da CID-10. Também foi realizada busca, pelos termos ‘congênit’, ‘malforma’, ‘displas’, ‘disrup’ e ‘genétic’, sendo considerados todos os códigos correspondentes.
As anomalias congênitas da CID-11 foram extraídas do capítulo 20, que compila os defeitos do desenvolvimento, bem como de todos os códigos relacionados aos erros inatos do metabolismo – anomalias congênitas por definição.
Utilizaram-se, ainda, as listas de anomalias de três modelos de vigilância: EUROCAT,(10),(11) ECLAMC (Latin American Collaborative Study of Congenital Malformations)(12),(13) e CREC (Costa Rican Birth Defects Register).(14) As redes ECLAMC e EUROCAT foram selecionadas por serem referência para a América-Latina e a Europa, respectivamente. O CREC foi escolhido por incluir um grande número de anomalias funcionais e vigiar a maior quantidade de anomalias congênitas fora do capítulo XVII da CID-10, comparado aos demais programas analisados.(13) O ECLAMC não possui lista específica de anomalias para vigilância, mas fornece um atlas de consulta, dotado de uma lista definida de condições.(13)
A partir da Orphanet,(8) foram aplicadas duas estratégias para extração das anomalias congênitas. Na primeira, entre as 35 classificações de doenças raras, seis atendiam ao conceito de anomalias congênitas: (i) Defeito de desenvolvimento raro durante a embriogênese; (ii) Malformações cardíacas raras; (iii) Erros inatos e raros do metabolismo; (iv) Doenças genéticas raras; (v) Desordens teratogênicas raras; e (vi) Anomalias cromossômicas classificadas por cromossomos. A segunda estratégia de busca procurou captar anomalias congênitas além dessas classificações, a partir do modelo de classificação hierárquica do tipo de desordens ( http://www.orpha.net/orphacom/cahiers/docs/GB/Orphanet_linearisation_rules.pdf ), considerando-se como anomalias congênitas as síndromes malformativas e anomalias morfológicas.
Os códigos das anomalias congênitas da CID-11 e da Orphanet foram mapeados, para correspondência com a CID-10, segundo orientação das próprias fontes. Da Orphanet, foram captados os códigos que mantinham correspondência exata com a CID-10. Posteriormente, as anomalias congênitas captadas foram compiladas em uma lista única, retirando-se as duplicatas.
Após processamento, a lista foi revisada manualmente (Bremm JM e Cardoso-dos-Santos AC), para exclusão das condições que não se enquadravam no conceito de anomalias congênitas. A lista obtida após essa revisão foi validada por duas geneticistas, especialistas na área (Sanseverino MTV e Schüler-Faccini L). Por fim, para classificação das anomalias na lista final, adotou-se o modelo proposto na CID-11, uma classificação mais atual e abrangente que a CID-10.
A Figura 1 apresenta as fontes consultadas e o processo para obtenção dos dados. Da CID-10, além dos 619 códigos do capítulo XVII, foram captados 509 mediante a busca por termos; feita a compilação de ambos os conjuntos e retiradas as duplicatas, chegou-se a 701 códigos. Na CID-11, o capítulo 20 (Anomalias do Desenvolvimento) conta com 1.311 códigos de anomalias congênitas, além dos 193 relacionados aos erros inatos do metabolismo, que, após correspondência com a CID-10, resultaram em 683 códigos únicos. Em relação aos modelos de vigilância, foram incluídos 638 códigos a partir do EUROCAT, 650 do ECLAMC e 731 do CREC, logo compilados e deduplicados, resultando em 767 códigos únicos.
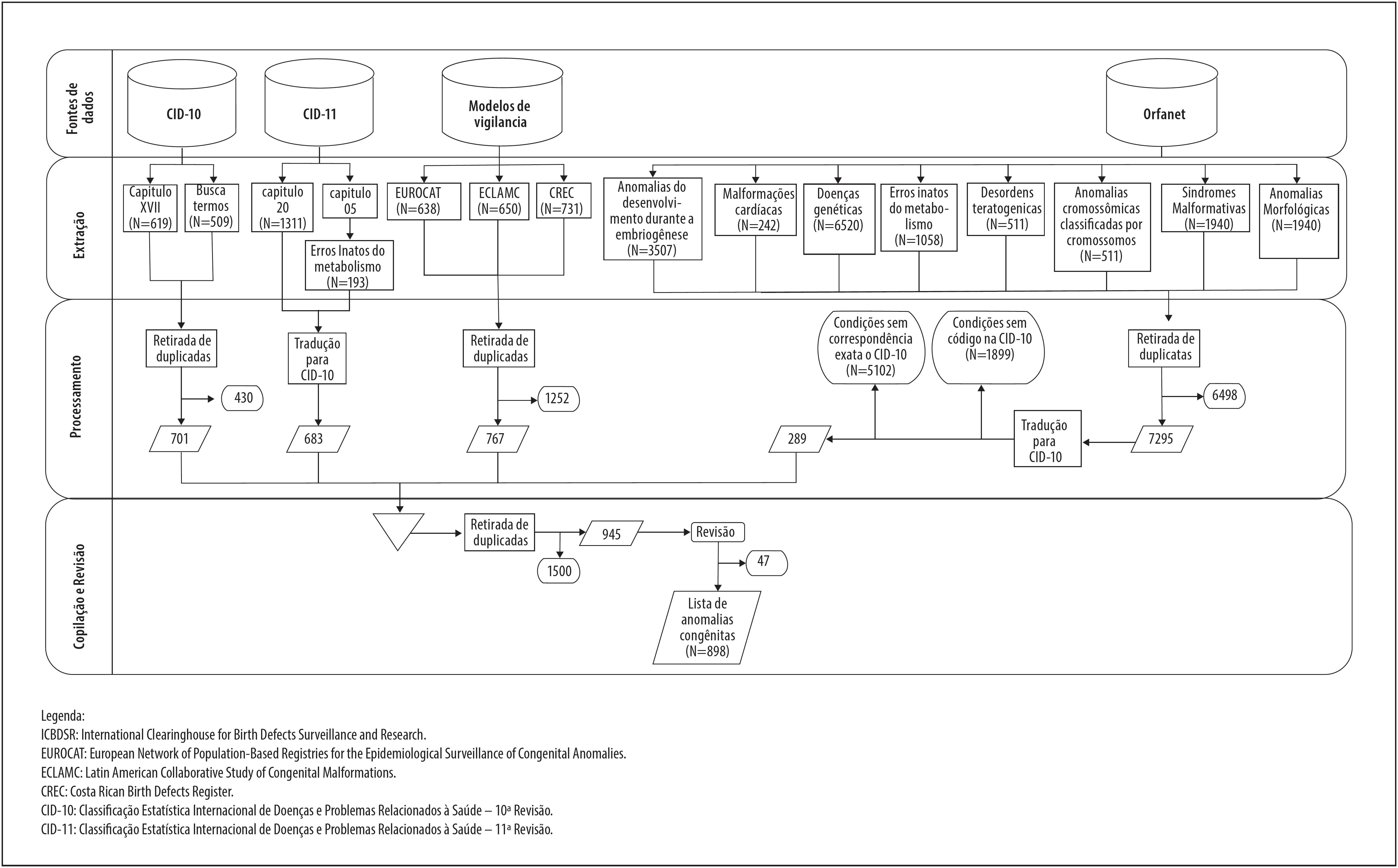
Entre as classificações da Orphanet, foram captados códigos referentes aos defeitos de desenvolvimento raros durante a embriogênese (3.507 códigos), malformações cardíacas raras (242), doenças genéticas raras (6.520), erros inatos e raros do metabolismo (1.058), desordens teratogênicas raras (39) e anomalias cromossômicas classificadas por cromossomos (511 códigos). Quanto ao tipo de desordens, as síndromes malformativas apresentaram 1.940 códigos, enquanto as anomalias morfológicas, 426. Uma vez compilados e de duplicados os códigos captados da Orphanet, foram excluídos 6.498. Dos 7.295 códigos traduzidos dessa base para a CID-10, apenas 289 (3,96% do total) apresentaram correspondência exata.
Em seguida, a lista de anomalias codificadas na CID-10, oriunda de diferentes fontes, foi compilada e deduplicada e 1.500 códigos foram excluídos. Os 945 códigos restantes passaram por revisão manual, sendo 47 definitivamente excluídos por não se enquadrarem no conceito de anomalias congênitas. Esse processo resultou em uma lista com 898 códigos de anomalias, logo classificados em 21 grupos distintos, de acordo com a proposta da CID-11 ( Figura 2 ).
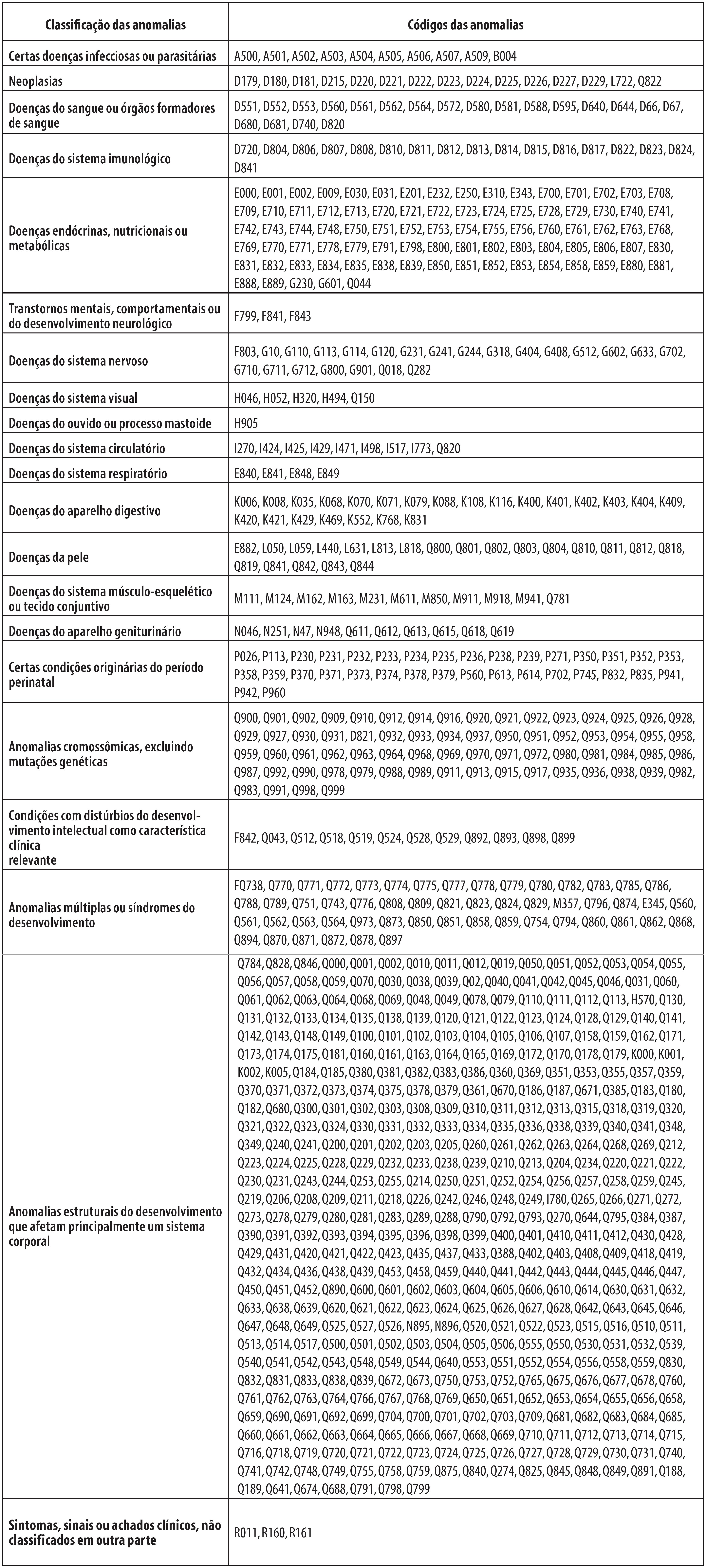
Figura 2 Classificação das anomalias congênitas de acordo com os capítulos da Classificação Estatística Internacional de Doenças e Problemas Relacionados à Saúde – Décima Primeira Revisão (CID-11))
A lista final, conclusiva deste trabalho, reuniu os 619 códigos do capítulo XVII da CID-10, além de 279 códigos de outros capítulos, dos quais 19 foram exclusivos da busca na CID-11, 72 dos modelos de vigilância, 79 da Orphanet e 36 da busca de termos na CID-10. Particularmente, 87 anomalias congênitas foram compartilhadas entre, pelo menos, duas dessas diferentes fontes ( Figura 3 ).

Discussão
Este trabalho propõe uma lista dotada de 898 anomalias com códigos correspondentes na CID-10, permitindo a instrumentalização do conceito de anomalias congênitas fornecido pela OMS. Comparada ao capítulo XVII da CID-10,(6) a lista elaborada incluiu 279 novas condições. O Sinasc está habilitado para inclusão dos códigos constantes do capítulo XVII da CID-10, tão somente, o que limita a vigilância de anomalias congênitas no Brasil.
Uma lista compreensiva de anomalias congênitas codificadas na CID-10 torna-se importante para a vigilância. Sua codificação é amplamente utilizada pelos profissionais de saúde, para registro de casos nos sistemas de informações. Entretanto, o mapeamento dos códigos foi um desafio. Em muitos casos, estes autores não encontraram a correspondência exata entre os códigos das fontes de dados utilizadas e os da CID-10, o que pode conduzir a erros. Para minimizar essa limitação, apenas anomalias de correspondência exata com a CID-10 foram selecionadas e, posteriormente, realizada conferência manual dos códigos, no sentido de refinar sua acurácia.
A habilitação de novos códigos da CID-10 para inclusão de anomalias congênitas nos sistemas de informações, especialmente no Sinasc, será de extrema relevância para a vigilância em saúde.(4) O volume atual não dispões de codificação para algumas condições, e o capítulo referente às anomalias congênitas na CID-10 (XVII) se restringe, basicamente, às anomalias estruturais. Embora tenham-se somado novas condições ao capítulo 20 na CID-11, tal medida não foi suficiente para contemplar a totalidade de condições passíveis de definição como anomalias congênitas. Os dados extraídos e analisados corroboram a declaração conjunta das duas maiores redes internacionais para vigilância de anomalias congênitas, EUROCAT e ICBDSR.(5)
Nesse contexto, a lista expandida, aqui proposta, fornece um escopo ampliado de anomalias congênitas, útil para os fins da vigilância dessas condições, seus objetivos e prioridades. Conclui-se que a incorporação de novas fontes de dados aproxima esta lista da totalidade de anomalias congênitas passíveis de registro no Brasil.