








Serviços Personalizados
Journal

Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Pan-Amazônica de Saúde
versão impressa ISSN 2176-6215versão On-line ISSN 2176-6223
Rev Pan-Amaz Saude v.1 n.1 Ananindeua mar. 2010
http://dx.doi.org/10.5123/S2176-62232010000100023
ARTIGO DE REVISÃO | REVIEW ARTICLE | ARTÍCULO DE REVISIÓN
Rabies pathogenesis update
Atualização sobre a patogênese da raiva
Actualización sobre la patogénesis de la rabia
Alan C. Jackson
Departments of Internal Medicine (Neurology) and of Medical Microbiology, University of Manitoba, Winnipeg, Manitoba, Canada
Endereço para correspondência
Correspondence
Dirección para correspondencia
ABSTRACT
Human rabies continues to be an important public health problem. Our understanding of the disease has been acquired from studies in experimental animal models. There are many unanswered questions in rabies pathogenesis, although there has been recent progress. Rabies virus-infected neurons may not function normally due to degenerative changes involving neuronal processes, including both dendrites and axons. Street rabies virus infection may not be cleared from the central nervous system because immune effectors cannot be delivered into brain tissues as a result of poor blood-brain barrier permeability. No effective therapy is available for human rabies. Therapeutic (induced) coma has failed repeatedly. An improved understanding of rabies pathogenesis may offer new insights for the development of novel therapies for human rabies.
Keywords: Virulence Factors; Rabies; Encephalitis, Viral.
RESUMO
A raiva humana continua sendo um problema de saúde pública. O nosso conhecimento acerca da doença tem sido construído por meio de estudos em modelos animais experimentais. Há muitas perguntas não respondidas envolvendo a patogênese da raiva, a despeito de termos observado um avanço nos últimos tempos. Os neurônios infectados pelo vírus da raiva podem não operar normalmente devido às mudanças degenerativas envolvendo processos neurais, incluindo dendrites e axônios. As infecções por vírus de rua não podem ser extirpadas do sistema nervoso central, uma vez que os efetores imunes não podem ser transportados aos tecidos do cérebro devido à pequena permeabilidade da barreira hematoencefálica. Não existe uma terapia eficaz contra a raiva humana. O coma terapêutico (induzido) tem falhado sistematicamente. Uma melhor compreensão da patogênese da raiva pode oferecer novas ideias para o desenvolvimento de modernas terapias contra a raiva humana.
Palavras-chave: Fatores de Virulência; Raiva; Encefalite Viral.
RESUMEN
La rabia humana sigue siendo un problema de salud pública. Nuestro conocimiento sobre esa enfermedad se ha construido a través de estudios en animales de experimentación. Hay muchas preguntas sin respuesta que rodean la patogénesis de la rabia, a pesar del avance observado en los últimos tiempos. Las neuronas infectadas con rabia no pueden funcionar normalmente debido a cambios degenerativos que afectan a procesos neurales, incluyendo a dendritas y axones. Las infecciones causadas por el virus natural de la rabia no pueden eliminarse del sistema nervioso central, ya que los efectores inmunológicos no pueden ser transportados a los tejidos del cerebro, debido a la permeabilidad de la barrera hematoencefálica. No existe una terapia eficaz contra la rabia humana. El estado de coma terapéutico (inducido) ha fracasado sistemáticamente. Una mejor comprensión de la patogénesis de la rabia puede aportar nuevas ideas para el desarrollo de terapias modernas contra la rabia humana.
Palabras clave: Factores de Virulencia; Rabia; Encefalitis Viral.
INTRODUCTION
Human rabies remains an important public health problem, with at least 55 thousand fatal cases per year51. Human rabies can be effectively prevented, but it remains an almost invariably fatal disease despite aggressive approaches to therapy14. Our basic understanding of rabies pathogenesis leaves many important questions unanswered. A better understanding of rabies pathogenesis may be helpful in making future advances in the therapy of human rabies. In this review, selected topics in the pathogenesis of rabies will be discussed with an emphasis on recent developments.
OVERVIEW OF RABIES PATHOGENESIS
Much of what is known about the pathogenesis of rabies has been learned from studies performed in animal models, usually in rodents infected with strains of fixed (laboratory adapted) rabies virus. There is a long and variable incubation period in human and animal rabies usually lasting 20 to 90 days, but in humans it may rarely last longer than one year44. After a bite from a rabid animal, saliva containing infectious rabies virus is inoculated into subcutaneous tissues and muscles. The best experimental animal studies to date examining the events that take place during the incubation period were performed in striped skunks using a Canadian isolate of street rabies virus obtained from skunk salivary glands4. Studies performed using reverse transcriptase-polymerase chain reaction (RT-PCR) amplification showed that when skunks were sacrificed 62 to 64 days post-inoculation, viral genomic RNA was frequently present in the inoculated muscle (found in four of nine skunks), but not in either spinal ganglia or the spinal cord. Immunohistochemical studies performed prior to the development of clinical disease showed evidence of infection of extrafusal muscle fibers and occasional fibrocytes at the site of inoculation. We can conclude that rabies virus is present at or near the site of the bite during most of the incubation period. The infection of muscle fibers may be a critical pathogenetic step for the virus to gain access to the peripheral nervous system.
Detailed pathogenesis studies of the early events in "natural" rabies models have not yet been performed in bats. Bats usually produce much more superficial bites than terrestrial vectors. There is a rich sensory and autonomic innervation of skin and subcutaneous tissues that becomes involved in a similar time course as with infections due to deeper-biting vectors. In North America most victims of rabies do not have a history of an animal bite, likely because they were not aware of the bite. In part, this is related to the small size of the bats, for example, silverhaired bats. The bat rabies virus variant most commonly responsible for human rabies in North America is associated with silver-haired/eastern pipistrelle bats37. Bat bites may produce lesions that appear quite trivial18. Experimental studies on the silver-haired bat virus indicate that the virus replicates well at lower than normal body temperatures (34o C) and has higher infectivity than coyote street virus in cell types present in the dermis, including fibroblasts and epithelial cells36. Hence, the silver-haired bat virus is likely well adapted for efficient local replication in the dermis, which could explain the success of this variant.
Rabies virus binds to nicotinic acetylcholine receptors at the neuromuscular junction32. The virus travels towards the central nervous system (CNS) within motor and sensory axons by retrograde fast axonal transport at a rate of 12-100 mm per day29,34,47. Rabies virus has been used as a neuroanatomical tracer in order to define circuits of synaptically-linked neurons in rodents and primates, and these studies have shown that axonal transport of rabies virus occurs exclusively in the retrograde direction45,27. When neurons are infected in the spinal cord, there is a subsequent spread from neuron to neuron within axons in the CNS by fast axonal transport along neuroanatomical connections. Many neuronal cell types are infected in a widespread distribution in the CNS; infection of nonneuronal cells occurs much less commonly. Brain infection results in behavioral changes, likely due to infection of neurons in limbic areas, and this facilitates transmission by biting in rabies vectors. There is a spread of rabies virus away from the CNS (centrifugal spread) along neuronal pathways, particularly involving the parasympathetic nervous system, which is responsible for infection of the salivary glands, skin (skin biopsy is a useful diagnostic test), heart, and a variety of other organs14,29. Infectious rabies virus is secreted into the saliva of rabies vectors, which is important for transmission to other hosts.
NEURONAL DYSFUNCTION AND DEATH IN RABIES VIRUS INFECTION
Natural rabies is normally characterized by severe neurologic signs and a fatal outcome. However, neuropathologic changes in the CNS are relatively mild, consisting of mild inflammation with little neuronal degeneration, supporting the concept that neuronal dysfunction, rather than neuronal cell death, plays an important role in producing the disease15,25. A variety of experimental studies of rabies virus infection have investigated potential abnormalities in neurotransmission involving acetylcholine13,48,6, serotonin3,2, and 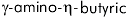 acid (GABA)30. Abnormalities of uncertain significance were found, but no fundamental defect was demonstrated that explains the neuronal dysfunction in rabies.
acid (GABA)30. Abnormalities of uncertain significance were found, but no fundamental defect was demonstrated that explains the neuronal dysfunction in rabies.
Dysfunction of ion channels has been shown in rabies virus – infected cultured mouse neuroblastoma NA cells with the whole-cell patch clamp technique12. The infection reduced the functional expression of voltage-dependent sodium channels and inward rectifier potassium channels, and there was a lower resting membrane potential reflecting membrane depolarization. There was no change in the expression of delayed rectifier potassium channels, indicating that nonselective dysfunction of ion channels had not occurred. The reduction in sodium channels and inward rectifier potassium channels could prevent infected neurons from firing action potentials and generating synaptic potentials, resulting in functional impairment.
Koprowski and co-workers28 have hypothesized that nitric oxide neurotoxicity may mediate neuronal dysfunction in rabies. Induction of inducible nitric oxide synthase mRNA28 and increased brain levels of nitric oxide10 have been demonstrated in rabies virus-infected rodents, but the significance of these findings remains uncertain. The role of nitric oxide in rabies pathogenesis needs further study. Excitoxicity has been studied in rabies virus-infected primary neuron cultures and in an experimental mouse model of rabies, but no importante role for excitoxicity in neuronal dysfunction or death was found49.
Using subtraction hybridization, Prosniak et al38 showed that infection of mice with fixed rabies virus results in downregulation of about 90% of genes in the normal brain by more than 4-fold. Only about 1.4% of genes became up-regulated, including genes involved in regulation of cell metabolism, protein synthesis, growth and differentiation. Using proteomic profiling of brain homogenates from mice infected with street (wild-type) rabies virus, Dhingra et al5 found that the levels of proteins involved in ion homeostasis were altered (H+ ATPase and Na+/K+ ATPase were up-regulated and Ca2+ ATPase was down-regulated). Also, they found down-regulation of proteins involved in docking and fusion of synaptic vesicles to the presynaptic membrane, a process relevant to synaptic physiology. These abnormalities could lead to neuronal dysfunction.
Neurotropic viruses may cause cell death by either apoptosis or necrosis9,1,7. Apoptosis depends on synthesis of macromolecules and requires energy, while necrosis is associated with energy failure. Each of these forms of cell death is associated with typical morphologic features. The challenge virus strain (CVS) of fixed rabies virus has been observed to induce apoptotic cell death in rat prostatic adenocarcinoma cells23, mouse neuroblastoma cells46, and mouse embryonic hippocampal neurons35. Prominent apoptotic death of neurons has been observed in the brains of mice of various ages inoculated intracerebrally with the CVS strain of fixed rabies virus23,46,19. However, neuronal death is not prominent after peripheral inoculation of adult animals43,15,39. Attenuated viruses are stronger inducers of neuronal apoptosis in cultured primary neurons and also in mice after peripheral inoculation than more virulent strains35,21. Contrary to previous reports26, we have recently provided strong evidence that neuronal apoptosis does not play an important role in human rabies20. Hence, in rabies virus infection there are complex mechanisms involved in cell death versus survival of neurons both in vitro and in animal models using different viral strains and routes of inoculation, but neuronal cell death is not prominent in natural rabies. In general, apoptosis is a host mechanism employed to limit viral spread and does not serve a fundamental role in the pathogenesis of rabies15.
During the prodromal period of rabies patients may experience pain, paresthesias, or itching at the site of the wound (often healed), which likely reflects involvement of local sensory (e.g., dorsal root) ganglia14. There is neuronal infection with inflammation and neuronal degeneration in dorsal root ganglia in human rabies and in most experimental models of rabies40. This neuronal degeneration is out of proportion with what is seen in the CNS of the mouse model after footpad inoculation with CVS40, and it is highly inflammatory, which suggests an immune-mediated process. The neuronal degeneration has neither the morphologic features of necrosis nor the morphologic or biochemical features of apoptosis. In gangliocytes there is an early 'axotomy response' and later the gangliocytes contain numerous autophagic compartments. At advanced stages of degeneration there are partially membrane-bound empty vacuoles in gangliocytes. Autophagy is an important mechanism involved in this degenerative neuronal process.
Li and colleagues33 have suggested that degeneration of neuronal processes and disruption of synaptic structures may form the basis for neuronal dysfunction in rabies virus infection. They showed severe destruction and disorganization of neuronal processes in silver stained hippocampal sections from mice infected intracerebrally with the pathogenic N2C strain of rabies virus. Our laboratory has recently examined morphological changes in neurons (with an emphasis on the structural integrity of neuronal processes) following hindlimb footpad inoculation of transgenic mice expressing yellow fluorescent protein (YFP) in a subpopulation of neurons, which facilitates visualization of the morphological details of dendrites, axons, and presynaptic nerve terminals43,8. In these mice, YFP expression is driven in a subpopulation of neurons using the thy1 vector lacking exon 3 and its flanking introns, and there are strong fluorescent signals in dendrites, axons, and presynaptic nerve terminals8. In this model, conventional histopathology showed mild inflammatory changes without significant degenerative neuronal changes. However, at late clinical time points concomitant with the development of severe clinical neurological disease, fluorescence microscopy showed marked abnormalities, especially beading and/or swelling involving dendrites and axons of layer V cortical pyramidal neurons, severe involvement of axons in the brainstem and the inferior cerebellar peduncle, and severe abnormalities affecting axons of cerebellar mossy fibers. The involvement of hippocampal pyramidal neurons was much less severe, perhaps because these neurons become infected much later after peripheral inoculation in this model22. The structural changes may take a period of time to develop perhaps because they are mediated by abnormal axoplasmic structural protein function. Toluidine bluestained resin sections and electron microscopy showed vacuolation in cortical neurons that corresponded to swollen mitochondria, and vacuolation in the neuropil of the cerebral cortex. Axonal swellings were observed. Vacuolation was also observed ultrastructurally in axons and in pre-synaptic nerve endings. These morphological changes are sufficient to explain the severe clinical disease and fatal outcome, and they provide strong evidence that the fundamental defect in rabies virus-infected neurons may involve neuronal processes, which is not apparent in routine histopathological studies.
BLOOD-BRAIN BARRIER IN RABIES
Hooper and colleagues10 compared mice infected with a lethal silver-haired bat rabies variant (SHBRV) with mice infected with an attenuated laboratory adapted strain (CVSF3) in which there is viral clearance without sequelae. They found that in both viral infections there was a strong virus-specific immune response in the periphery of the host. However, invasion of the CNS tissues by immune cells was reduced with the SHBRV due to an inability to enhance blood-brain barrier permeability, resulting in failure of viral clearance42. Mice with an SJL background are less susceptible to lethal infection with rabies virus and have a greater capacity to mediate CNS inflammatory responses. SHBRV infection in these mice is associated with the induction of greater blood-brain barrier permeability and CNS inflammation, resulting in greater viral clearance and improved survival41. Administration of a steroid hormone in these mice resulted in reduced blood-brain barrier changes and resulted in greater mortality41. Hence, blood-brain barrier permeability is likely of paramount importance for viral clearance in rabies and therapeutic approaches should be taken to enhance this permeability. Therapies that promote closure the blood-brain barrier, such as the administration of corticosteroids, should be carefully avoided in the management of human rabies patients.
APPROACHES TO THE THERAPY OF HUMAN RABIES
There is no established therapy for human patients with rabies. An approach to the management of human rabies was recently published24. This article recommended using a combination of therapies and discussed the pros and cons of using a variety of specific agents. In 2004, a 15-year-old patient survived rabies who had not received rabies vaccine prior to the onset of clinical disease50. She was bitten by a bat on a finger and did not seek medical attention or receive treatment at that time. About one month after the bite, she developed typical clinical features of rabies, and neutralizing anti-rabies virus antibodies were detected in her sera and cerebrospinal fluid. Her therapy included therapeutic (induced) coma using intravenous midazolam for seven days; a burst-suppression pattern on her electroencephalogram was maintained and supplemental phenobarbital was given. She also received therapy with ketamine and antiviral therapy, including ribavirin and amantadine. She improved and was discharged from the hospital with neurologic deficits, but she subsequently had progressive neurologic improvement11. This is the first documented survivor who had not received rabies vaccine prior to the onset of clinical rabies. As discussed in the accompanying editorial, it is unknown if therapy with one or more specific agents played an important role in the favorable outcome of this patient16. However, since that time, there have been at least 13 known cases in which the main components of this approach (the "Milwaukee" protocol) have been used, and fatal outcomes have resulted17. The induction of coma per se has not been shown to be useful in the management of infectious diseases of the nervous system, and there is no evidence supporting this approach in rabies or other viral encephalitises. Hence, therapeutic coma should not become a routine therapy for the management of rabies.
The development of neutralizing anti-rabies virus antibodies early in the patient's clinical course, which probably occurs in less than 20% of all patients with rabies, likely contributed to a favorable outcome. Bat rabies viruses may be less neurovirulent than canine or other variants that are responsible for most human cases of rabies31, and rabies due to canine rabies virus variants may have a worse outcome than cases caused by bat rabies variants. Finally, most survivors of rabies have shown neutralizing anti-rabies virus antibodies in sera and cerebrospinal fluid, but other diagnostic laboratory tests are usually negative for rabies virus antigen and RNA in fluids and tissues (brain tissues are not tested). This may be due to effective viral clearance.
CONCLUSIONS
The bases for neuronal dysfunction in rabies are complex, but they may involve degenerative changes involving neuronal processes such as dendrites and axons. For reasons that are unclear, neuronal injury is greater in dorsal root ganglia than in CNS neurons. Permeability of the blood-brain barrier to immune effectors is important for viral clearance and recovery from rabies. No effective therapy for human rabies is available. Hopefully, an improved understanding of rabies pathogenesis will lead to the development of novel therapies for human rabies.
REFERENCES
1 Allsopp TE, Fazakerley JK. Altruistic cell suicide and the specialized case of the virus-infected nervous system. Trends Neurosci. 2000 Jul;23(7):284-90. DOI:10.1016/S0166-2236(00)01591-5 [ Links ]
2 Bouzamondo E, Ladogana A, Tsiang H. Alteration of potassium-evoked 5-HT release from virus-infected rat cortical synaptosomes. Neuro Report. 1993 May;4(5):555-8. [ Links ]
3 Ceccaldi PE, Fillion MP, Ermine A, Tsiang H, Fillion G. Rabies virus selectively alters 5-HT1 receptor subtypes in rat brain. Eur J Pharmacol. 1993 Apr;245(2):129-38. [ Links ]
4 Charlton KM, Nadin-Davis S, Casey GA, Wandeler AI. The long incubation period in rabies: delayed progression of infection in muscle at the site of exposure. Acta Neuropathol. 1997 Jul;94(1):73-7. [ Links ]
5 Dhingra V, Li X, Liu Y, Fu ZF. Proteomic profiling reveals that rabies virus infection results in differential expression of host proteins involved in ion homeostasis and synaptic physiology in the central nervous system. J Neurovirol. 2007 Apr;13(2):107-17. [ Links ]
6 Dumrongphol H, Srikiatkhachorn A, Hemachudha T, Kotchabhakdi N, Govitrapong P. Alteration of muscarinic acetylcholine receptors in rabies viral-infected dog brains. J Neurol Sci. 1996 Apr;137(1):1-6. DOI:10.1016/0022-510X(95)00275-7 [ Links ]
7 Fazakerley JK, Allsopp TE. Programmed cell death in virus infections of the nervous system. Curr Top Microbiol Immunol. 2001;253:95-119.
8 Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000 Oct;28(1):41-51. [ Links ]
9 Griffin DE, Hardwick JM. Perspective: virus infections and the death of neurons. Trends Microbiol. 1999 Apr;7(4):155-60. DOI:10.1016/S0966-842X(99)01470-5 [ Links ]
10 Hooper DC, Ohnishi ST, Kean R, Numagami Y, Dietzschold B, Koprowski H. Local nitric oxide production in viral and autoimmune diseases of the central nervous system. Proc Natl Acad Sci U S A. 1995 Jun;92(12):5312-6. [ Links ]
11 Hu WT, Willoughby RE Jr, Dhonau H, Mack KJ. Longterm follow-up after treatment of rabies by induction of coma (Letter). N Engl J Med. 2007 Aug;357(9):945-6.
12 Iwata M, Komori S, Unno T, Minamoto N, Ohashi H. Modification of membrane currents in mouse neuroblastoma cells following infection with rabies virus. Br J Pharmacol. 1999 Apr;126(8):1691-8. DOI:10.1038/sj.bjp.0702473 [ Links ]
13 Jackson AC. Cholinergic system in experimental rabies in mice. Acta Virol. 1993 Dec;37(6):502-8. [ Links ]
14 Jackson AC. Human disease. In: Jackson AC, Wunner WH, editors. Rabies. 2nd ed. London: Elsevier Academic Press; 2007. p. 309-40.
15 Jackson AC. Pathogenesis. In: Jackson AC, Wunner WH, editors. Rabies, 2nd ed. London: Elsevier Academic Press; 2007. p. 341-81.
16 Jackson AC. Recovery from rabies. N Engl J Med. 2005 Jun;352(24):2549-50.
17 Jackson AC. Update on rabies diagnosis and treatment. Curr Infect Dis Rep. 2009 Jul;11(4):296- 301. [ Links ]
18 Jackson AC, Fenton MB. Human rabies and bat bites. Lancet. 2001 May;357(9269):1714.
19 Jackson AC, Park H. Apoptotic cell death in experimental rabies in suckling mice. Acta Neuropathol. 1998 Feb;95(2):159-64. [ Links ]
20 Jackson AC, Randle E, Lawrance G, Rossiter JP. Neuronal apoptosis does not play an important role in human rabies encephalitis. J Neurovirol. 2008 Oct;14(5):368-75. DOI:10.1080/13550280802216502 [ Links ]
21 Jackson AC, Rasalingam P, Weli SC. Comparative pathogenesis of recombinant rabies vaccine strain SAD-L16 and SAD-D29 with replacement of Arg333 in the glycoprotein after peripheral inoculation of neonatal mice: less neurovirulent strain is a stronger inducer of neuronal apoptosis. Acta Neuropathol. 2006 Apr;111(4):372-8. DOI:10.1007/s00401-005-0006-z [ Links ]
22 Jackson AC, Reimer DL. Pathogenesis of experimental rabies in mice: an immunohistochemical study. Acta Neuropathol. 1989;78(2):159-65. [ Links ]
23 Jackson AC, Rossiter JP. Apoptosis plays an important role in experimental rabies virus infection. J Virol. 1997 Jul;71(7):5603-7. [ Links ]
24 Jackson AC, Warrell MJ, Rupprecht CE, Ertl HC, Dietzschold B, O'Reilly M, et al. Management of rabies in humans. Clin Infect Dis. 2003 Jan;36(1):60-3. [ Links ]
25 Jackson AC, Wunner WH. Rabies. 2nd ed. London: Elsevier Academic Press; 2007.
26 Juntrakul S, Ruangvejvorachai P, Shuangshoti S, Wacharapluesadee S, Hemachudha T. Mechanisms of escape phenomenon of spinal cord and brainstem in human rabies. BMC Infect Dis. 2005 Nov;5(1):104.
27 Kelly RM, Strick PL. Rabies as a transneuronal tracer of circuits in the central nervous system. J Neurosci Methods. 2000 Nov;103(1):63-71. DOI:10.1016/S0165-0270(00)00296-X [ Links ]
28 Koprowski H, Zheng YM, Heber-Katz E, Fraser N, Rorke L, Fu ZF, et al. In vivo expression of inducible nitric oxide synthase in experimentally induced neurologic diseases. Proc Natl Acad Sci U S A. 1993 Apr;90(7):3024-7. Erratum in: Proc Natl Acad Sci U S A. 1993 Jun;90(11):5378. [ Links ]
29 Kucera P, Dolivo M, Coulon P, Flamand A. Pathways of the early propagation of virulent and avirulent rabies strains from the eye to the brain. J Virol. 1985 Jul;55(1):158-62. [ Links ]
30 Ladogana A, Bouzamondo E, Pocchiari M, Tsiang H. Modification of tritiated γ-amino-η-butyric acid transport in rabies virus-infected primary cortical cultures. J Gen Virol. 1994 Mar;75(Pt 3):623-7. [ Links ]
31 Lafon M. Bat rabies: the Achilles heel of a viral killer? Lancet. 2005 Sep;366(9489):876-7.
32 Lentz TL, Burrage TG, Smith AL, Crick J, Tignor GH. Is the acetylcholine receptor a rabies virus receptor? Science. 1982 Jan;215(4529):182-4. [ Links ]
33 Li XQ, Sarmento L, Fu ZF. Degeneration of neuronal processes after infection with pathogenic, but not attenuated, rabies viruses. J Virol. 2005 Aug;79(15):10063-8. DOI:10.1128/JVI.79.15.10063–10068.2005 [ Links ]
34 Lycke E, Tsiang H. Rabies virus infection of cultured rat sensory neurons. J Virol. 1987 Sep;61(9):2733-41. [ Links ]
35 Morimoto K, Hooper DC, Spitsin S, Koprowski H, Dietzschold B. Pathogenicity of different rabies virus variants inversely correlates with apoptosis and rabies virus glycoprotein expression in infected primary neuron cultures. J Virol. 1999 Jan;73(1):510-8. [ Links ]
36 Morimoto K, Patel M, Corisdeo S, Hooper DC, Fu ZF, Rupprecht CE, et al. Characterization of a unique variant of bat rabies virus responsible for newly emerging human cases in North America. Proc Natl Acad Sci U S A. 1996 May;93(11):5653-8. [ Links ]
37 Noah DL, Drenzek CL, Smith JS, Krebs JW, Orciari L, Shaddock J, et al. Epidemiology of human rabies in the United States, 1980 to 1996. Ann Intern Med. 1998 Jun;128(11):922-30. [ Links ]
38 Prosniak M, Hooper DC, Dietzschold B, Koprowski H. Effect of rabies virus infection on gene expression in mouse brain. Proc Natl Acad Sci U S A. 2001 Feb;98(5):2758-63. DOI: 10.1073/pnas.051630298 [ Links ]
39 Reid JE, Jackson AC. Experimental rabies virus infection in Artibeus jamaicensis bats with CVS-24 variants. J Neurovirol. 2001 Dec;7(6):511-7. DOI: 10.1080/135502801753248097 [ Links ]
40 Rossiter JP, Hsu L, Jackson AC. Selective vulnerability of dorsal root ganglia neurons in experimental rabies after peripheral inoculation of CVS-11 in adult mice. Acta Neuropathol. 2009 Aug;118(2):249-59. [ Links ]
41 Roy A, Hooper DC. Lethal silver-haired bat rabies virus infection can be prevented by opening the blood-brain barrier. J Virol. 2007 Aug;81(15):7993-8. DOI:10.1128/JVI.00710-07 [ Links ]
42 Roy A, Phares TW, Koprowski H, Hooper DC. Failure to open the blood-brain barrier and deliver immune effectors to central nervous system tissues leads to the lethal outcome of silver-haired bat rabies virus infection. J Virol. 2007 Feb;81(3):1110-8. DOI: 10.1128/JVI.01964-06 [ Links ]
43 Scott CA, Rossiter JP, Andrew RD, Jackson AC. Structural abnormalities in neurons are sufficient to explain the clinical disease and fatal outcome of experimental rabies in yellow fluorescent protein-expressing transgenic mice. J Virol. 2008 Jan;82(1):513-21. DOI:10.1128/JVI.01677-07 [ Links ]
44 Smith JS, Fishbein DB, Rupprecht CE, Clark K. Unexplained rabies in three immigrants in the United States. A virologic investigation. N Engl J Med. 1991 Jan;324(4):205-11. [ Links ]
45 Tang Y, Rampin O, Giuliano F, Ugolini G. Spinal and brain circuits to motoneurons of the bulbospongiosus muscle: retrograde transneuronal tracing with rabies virus. J Comp Neurol. 1999 Nov;414(2):167-92. [ Links ]
46 Theerasurakarn S, Ubol S. Apoptosis induction in brain during the fixed strain of rabies virus infection correlates with onset and severity of illness. J Neurovirol. 1998 Aug;4(4):407-14. [ Links ]
47 Tsiang H, Ceccaldi PE, Lycke E. Rabies virus infection and transport in human sensory dorsal root ganglia neurons. J Gen Virol. 1991 May;72(Pt 5):1191-4.
48 Tsiang H. Neuronal function impairment in rabies-infected rat brain. J Gen Virol. 1982 Aug;61(Pt 2):277-81. [ Links ]
49 Weli SC, Scott CA, Ward CA, Jackson AC. Rabies virus infection of primary neuronal cultures and adult mice: failure to demonstrate evidence of excitotoxicity. J Virol. 2006 Oct;80(20):10270-3. DOI:10.1128/JVI.01272-06 [ Links ]
50 Willoughby RE Jr, Tieves KS, Hoffman GM, Ghanayem NS, Amlie-Lefond CM, Schwabe MJ, et al. Survival after treatment of rabies with induction of coma. N Engl J Med. 2005 Jun;352(24):2508-14. [ Links ]
51 World Health Organization. WHO expert consultation on rabies: first report. Geneva; 2005. p. 1-88.
 Correspondência/Correspondence/Correspondencia:
Correspondência/Correspondence/Correspondencia:
Alan C. Jackson
Health Sciences Centre
GF-543, 820 Sherbrook Street
Winnipeg, Manitoba R3A 1R9 Canada
Phone: 204-787-1578 Fax: 204-787-1486
E-mail: ajackson2@hsc.mb.ca
Recebido em/Received/Recibido en: 21/06/2009
Aceito em/Accepted/Aceito en: 02/10/2009