








Serviços Personalizados
Journal

Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Pan-Amazônica de Saúde
versão impressa ISSN 2176-6215versão On-line ISSN 2176-6223
Rev Pan-Amaz Saude v.1 n.1 Ananindeua mar. 2010
http://dx.doi.org/10.5123/S2176-62232010000100023
Atualização sobre a patogênese da raiva
Alan C. Jackson
Departments of Internal Medicine (Neurology) and of Medical Microbiology, University of Manitoba, Winnipeg, Manitoba, Canada
Endereço para correspondência
Correspondence
Dirección para correspondencia
Título original: Rabies pathogenesis update. Traduzido por: André Diniz.
RESUMO
A raiva humana continua sendo um problema de saúde pública. O nosso conhecimento acerca da doença tem sido construído por meio de estudos em modelos animais experimentais. Há muitas perguntas não respondidas envolvendo a patogênese da raiva, a despeito de termos observado um avanço nos últimos tempos. Os neurônios infectados pelo vírus da raiva podem não operar normalmente devido às mudanças degenerativas envolvendo processos neurais, incluindo dendrites e axônios. As infecções por vírus de rua não podem ser extirpadas do sistema nervoso central, uma vez que os efetores imunes não podem ser transportados aos tecidos do cérebro devido à pequena permeabilidade da barreira hematoencefálica. Não existe uma terapia eficaz contra a raiva humana. O coma terapêutico (induzido) tem falhado sistematicamente. Uma melhor compreensão da patogênese da raiva pode oferecer novas ideias para o desenvolvimento de modernas terapias contra a raiva humana.
Palavras-chave: Fatores de Virulência; Raiva; Encefalite Viral.
INTRODUÇÃO
A raiva humana permanece sendo um importante problema de saúde pública, responsável por no mínimo de 55.000 casos fatais por ano51. A raiva humana pode ser prevenida de forma eficaz, porém permanece sendo uma doença quase que invariavelmente fatal, a despeito da existência de condutas terapêuticas agressivas14. O nosso conhecimento básico acerca da patogênese da raiva pode deixar diversas questões importantes sem resposta. Uma melhor compreensão da patogênese da raiva pode ser muito útil para que se consiga avançar na terapia para raiva humana no futuro. Nesta revisão, alguns assuntos selecionados sobre a patogênese da raiva serão discutidos, com ênfase em recentes descobertas.
REVISÃO DA PATOGÊNESE DA RAIVA
Muito do que é conhecido acerca da patogênese da raiva foi descoberto a partir de estudos efetuados em modelos animais, geralmente em roedores infectados com cepas de vírus fixos da raiva (adaptados em laboratório). O período de incubação da raiva nas espécies humana e animal é longo e variável, normalmente durando entre 20 e 90 dias; entretanto, nos seres humanos este período pode, em raros casos, durar mais de um ano44. Após a mordida efetuada pelo animal infectado, a saliva contendo o vírus da raiva é inoculada nos tecidos e músculos subcutâneos. Os estudos experimentais que melhor examinaram os eventos que ocorrem durante o período de incubação até o presente momento foram executados em gambás listrados americanos (Mephitis mephitis), utilizando-se um isolado canadense de vírus rábico de rua obtido de suas glândulas salivares4. Os estudos realizados utilizando-se amplificação por transcriptase reversa -reação em cadeia de polimerase (RT-PCR) mostraram que, quando os gambás foram sacrificados 62 - 64 dias pós-inoculação, o RNA genômico viral era frequentemente encontrado no músculo inoculado (em 4 de 9 gambás), porém não era encontrado nem nos gânglios espinhais nem na medula espinhal. Estudos imuno-histoquímicos realizados anteriormente ao desenvolvimento da doença apresentaram evidências de infecção em fibras musculares extrafusais e fibrócitos ocasionais no ponto de inoculação. Podemos concluir que o vírus da raiva está presente no local da mordida ou em local próximo durante a maior parte do período de incubação. A infecção das fibras musculares pode ser uma fase patogenética crítica para o acesso do vírus ao sistema nervoso periférico.
Estudos detalhados acerca dos eventos iniciais da patogênese em modelos "naturais" de raiva não têm sido realizados em morcegos. Os morcegos normalmente são responsáveis por mais mordidas superficiais do que vetores terrestres. Há uma rica inervação sensorial e autonômica na pele e em tecidos subcutâneos, que é envolvida em um período de tempo similar ao das infecções causadas por vetores que provocam mordidas mais profundas. Na América do Norte, a maior parte das vítimas de raiva não apresenta histórico de mordida por animais, provavelmente devido ao fato de não as perceberem. Em parte, este fato é relacionado ao tamanho diminuto das mordidas - como exemplo, podemos citar os morcegos de pelos prateados (silver-haired bats). A variante de vírus rábico de morcego mais comumente responsável pela raiva humana na América do Norte está associada aos morcegos de pelo prateado/morcegos orientais pipistrellus37. As mordidas de morcego podem ocasionar lesões que parecem bem comuns18. Estudos experimentais com vírus de morcegos de pelos prateados indicam que ele se replica bem em temperaturas corporais mais baixas do que a normal (34o C) e que tem maior infectividade que a variante "de rua" de coyote em tipos de células presentes na derme, incluindo aqui os fibroblastos e as células epiteliais36. Por isso, o vírus de morcegos de pelos prateados é provavelmente bem adaptado para realizar uma eficiente replicação local na derme, o que poderia explicar o sucesso desta variante.
O vírus da raiva se liga a receptores nicotínicos de acetilcolina na junção neuromuscular32. O vírus se desloca para o sistema nervoso central (SNC) dentro de axônios motores e axônios sensoriais por meio do transporte axonal retrógrado rápido a uma taxa de 12 -100 mm por dia29,34,47. O vírus da raiva tem sido utilizado como marcador neuroanatômico a fim de definir os circuitos de neurônios ligados sinapticamente em roedores e primatas. Estes estudos têm demonstrado que o transporte axonal do vírus da raiva ocorre exclusivamente no sentido retrógrado45,27. Quando os neurônios são infectados na medula espinhal, ocorre uma posterior disseminação neurônio a neurônio dentro dos axônios no SNC por meio do transporte axonal rápido em conexões neuroanatômicas. Muitos tipos de células neuronais são infectados em uma ampla disseminação no SNC; a infecção de células não-neuronais ocorre em uma frequência bem menor. A infecção cerebral ocasiona mudanças de comportamento, provavelmente devido à infecção de neurônios em áreas límbicas, o que facilita a transmissão por mordidas em vetores da raiva. Há uma disseminação do vírus da raiva distante do SNC (d issem i na ção cen trífug a) por via s n eu ron ai s, parti cu l arm ente envol vendo o sistema nervoso parassimpático, que é responsável pela infecção das glândulas salivares, da pele (biópsia da pele é um exame diagnóstico útil), do coração e de uma variedade de outros órgãos14,29. O vírus da raiva infeccioso é secretado na saliva de vetores da raiva, o que se configura importante para a transmissão para outros reservatórios.
DISFUNÇÃO NEURONAL E MORTE EM INFECÇÕES PELO VÍRUS DA RAIVA
A raiva natural é normalmente caracterizada por sinais neurológicos graves e resultados fatais. Entretanto, as mudanças neuropatológicas no SNC são relativamente leves, consistindo em inflamação leve, acompanhada de pequena degeneração neuronal, corroborando o conceito de que a disfunção neuronal, mais do que a morte da célula neuronal, desempenha um papel importante na produção da doença15,25. Diversos estudos experimentais sobre a infecção pelo vírus da raiva têm investigado potenciais an orm alid ades na neu rotransmi ssão envolvendo acetilcolina13,48,6, serotonina3,2, e ácido 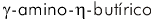 (GABA)30. Foram encontradas anormalidades de importância controversa, porém nenhum defeito fundamental que explicasse a disfunção neuronal na raiva foi demonstrado.
(GABA)30. Foram encontradas anormalidades de importância controversa, porém nenhum defeito fundamental que explicasse a disfunção neuronal na raiva foi demonstrado.
A disfunção de canais de íons já foi demonstrada no vírus da raiva em cultura de células de neuroblastoma de camundongos (NA) infectadas, com o uso da técnica de Patch-Clamp de célula inteira12. A infecção reduziu a expressão funcional dos canais de sódio voltagem-dependentes e de canais de potássio corretores do fluxo de internalização. Houve também uma redução no potencial de membrana restante, refletindo sua despolarização. Não houve alteração na expressão dos canais de potássio de retificação tardia, indicando que a disfunção não seletiva de canais iónicos não havia ocorrido. A redução dos canais de sódio e dos canais de potássio corretores do fluxo de internalização poderia impedir neurónios infectados de utilizar seus potenciais de ação e de gerar potenciais sinápticos, o que resultaria em prejuízo funcional.
Koprowski e colaboradores28 levantaram a hipótese de que a neurotoxicidade do óxido nítrico poderia mediar a disfunção neuronal na raiva. A indução de mRNA óxido nítrico sintetase indutível28 e o aumento dos níveis de óxido nítrico no cérebro10 já foram demonstrados em roedores infectados pelo vírus da raiva, porém a relevância destes achados permanece incerta. É necessário que se estude mais o papel do óxido nítrico na patogênese da raiva. A exocitoxicidade foi estudada em culturas primárias de neurónios infectados pelo vírus da raiva e em um modelo experimental de camundongo infectado pelo vírus da raiva, porém não foi constatado um papel relevante para a exocitoxicidade na disfunção neuronal ou na morte.
Usando hibridização subtrativa, Prosniak et al25 demonstraram que a infecção de camundongos por vírus fixo da raiva resulta em uma diminuição da regulação da expressão gênica de cerca de 90% em comparação ao cérebro normal em mais de quatro vezes. Cerca de 1,4% dos genes, apenas, foram positivamente regulados, incluindo os genes envolvidos na regulação do metabolismo celular, na síntese de proteínas, no crescimento e diferenciação. Usando de análise de perfil proteómico de homogeneizados de cérebro de camundongos infectados por vírus da raiva "de rua" (selvagem), Dhingra et al5 perceberam que os níveis de proteínas envolvidas na homeostase iónica estavam alterados (H+ ATPase e Na+/K+ ATPase foram positivamente regulados, e Ca2+ ATPase foi negativamente regulado). Eles também notaram uma regulação negativa de proteínas envolvidas no ancoramento e fusão de vesículas sinápticas com a membrana plasmática pressináptica, um processo importante para a fisiologia sináptica. Essas anormalidades podem levar à disfunção neuronal.
Vírus neurotrópicos podem causar a morte celular tanto por apoptose como por necrose9,1,7. A apoptose depende da síntese de macromoléculas e requer energia, enquanto que a necrose está associada a uma falência energética. Cada um desses tipos de morte celular está associado a características morfológicas típicas. Observou-se que a cepa viral de desafio (CVS) de vírus fixo da raiva induziu a morte celular por apoptose em células de adenocarcinoma de próstata de camundongos23, em células de neuroblastoma de camundongo46 e em neurônios embrionários do hipocampo de camundongos35. A morte de neurônios por apoptose foi observada principalmente em cérebros de camundongos de diferentes idades, inoculados via intracerebral com a cepa CVS do vírus rábico fixo23,46,19. No entanto, a morte neuronal não é proeminente após a inoculação periférica em animais adultos43,15,39. Os vírus atenuados são melhores indutores de apoptose neuronal em culturas de neurônios primários, bem como em camundongos após inoculação periférica, do que a maior parte das cepas virulentas35,21. Contrariamente ao já encontrado em estudos anteriores26, recentemente fornecemos fortes evidências de que a apoptose neuronal não exerce um papel importante no que diz respeito à raiva humana20. Por isso, em infecções causadas pelo vírus da raiva há mecanismos complexos envolvidos na morte de células em relação à sobrevivência de neurônios tanto in vitro como em modelos animais usando diferentes cepas virais e vias de inoculação; no entanto, a morte de células neuronais não se configura proeminente na raiva natural. Em geral, a apoptose é um mecanismo do hospedeiro utilizado para limitar a disseminação viral, e não exerce papel fundamental na patogênese da raiva15.
Durante o período prodrômico da raiva, os pacientes podem experimentar dor, parestesia ou prurido no lugar da ferida (geralmente já curada), que provavelmente reflete o envolvimento de gânglios sensitivos locais (por exemplo, da raiz nervosa espinhal)14. Há uma infecção neuronal, acompanhada de inflamação e degeneração neuronal nos gânglios, na raiva humana e na maioria dos modelos experimentais de raiva40. Esta degeneração neuronal é desproporcional ao que é visto no SNC do modelo murino após a inoculação plantar com o CVS40, e é também altamente inflamatória, o que sugere um processo imunomediado. A degeneração neuronal não apresenta nem as características morfológicas da necrose nem a características morfológicas ou bioquímicas da apoptose. Nos gangliócitos ocorre inicialmente uma "resposta axônica", e, posteriormente, os gangliócitos apresentam muitos compartimentos autofágicos. Em estágios avançados de degeneração, aparecem vacúolos vazios parcialmente limitados por membrana nos gangliócitos. A autofagia é um relevante mecanismo envolvido neste processo degenativo neuronal.
Li e colaboradores33 sugeriram que a degeneração de processos neuronais e a ruptura das estruturas sinápticas podem formar a base para a disfunção neuronal na infecção por vírus da raiva. Eles demonstraram danos graves e a desorganização dos processos neuronais em cortes do hipocampo de camundongos corados com prata, em animais infectados via intracerebral com a cepa patogênica N2C do vírus da raiva. Recentemente nosso laboratório examinou mudanças morfológicas em neurônios (com ênfase na integridade estrutural dos processos neuronais) após a inoculação na superfície plantar da pata traseira de camundongos transgênicos expressando proteína fluorescente amarela em uma subpopulação de neurônios, o que facilita a visualização de detalhes morfológicos dos dendritos, axônios e terminais nervosos pressinápticos43,8. Nesses camundongos, a expressão de proteína fluorescente amarela é dirigida a uma subpopulação de neurônio usando o vetor thyl desprovido de éxon 3 e suas regiões "flanqueadas", e observam-se fortes sinais fluorescentes em dendritos, axônios e terminais nervosos pressinápticos8. Neste modelo, a histopatologia convencional apresentou leves alterações inflamatórias, sem alterações significantes na degeneração neuronal. Entretanto, em momentos clínicos posteriores, concomitantemente com o desenvolvimento de doença clínica neurológica, a microscopia fluorescente apontou anormalidades acentuadas, principalmente o aparecimento de edema que envolviam dendrites e axônios de neurônios piramidais da quinta camada cortical, um severo envolvimento de axônios no tronco encefálico e no pendúnculo cerebelar inferior e graves anormalidades que afetavam os axônios de fibras nervosas cerebelares. O envolvimento de neurônios piramidais de hipocampo foi bem menos severo, talvez porque esses neurônios tenham sido infectados bem depois da inoculação periférica neste modelo22. As mudanças estruturais podem levar um certo tempo para se desenvolverem porque são mediadas por uma função proteica axoplásmica estrutural axonal. Secções em resina e coradas com azul de toluidina, bem como a microscopia eletrônica mostraram uma vacuolização em neurônios corticais que correspondiam a mitocôndrias edemaciadas e vacuolização nos neurópilos do córtex cerebral. Edemas axonais foram observados. Ultraestruturalmente, também foi observada vacuolização em axônios e em terminais nervosos pressinápticos. Estas alterações morfológicas são suficientes para explicar a doença clínica severa e o desfecho fatal. Elas também fornecem fortes evidências de que a principal deficiência de neurônios infectados pelo vírus da raiva envolveria processos neuronais, o que não se configura aparente em análises histopatológicas de rotina.
BARREIRA HEMATOENCEFÁLICA NA RAIVA
Hooper e colaboradores10 compararam camundongos infectados com uma variante letal de raiva em morcegos de pelos prateados (SHBRV) a camundongos infectados com uma cepa adaptada, atenuada em laboratório (CVS-F3) na qual se podia observar a eliminação viral sem a existência de sequelas. Eles descobriram que em ambas as infecções virais havia uma forte resposta imune-específica ao vírus na periferia do hospedeiro. Contudo, a invasão de células imunes nos tecidos do SNC foi diminuída com a SHRBV devido a uma incapacidade de aumento da permeabilidade da barreira hematoencefálica, o que, por sua vez, resultou em uma falha no clearance viral42. Camundongos de uma linhagem SJL são menos suscetíveis à infecção letal por vírus da raiva, e apresentam uma maior capacidade de mediar respostas inflamatórias no SNC. As infecções por SHBRV nestes camundongos está associada à indução de uma maior permeabilidade da barreira hematoencefálica e à inflamação do SNC, o que resulta em uma eliminação viral maior e uma taxa de sobrevivência maior41. A aplicação de hormônio esteroide nestes camundongos resultou em trocas reduzidas na barreira hematoencefálica e que resultou em uma maior mortalidade41. Por isso, a permeabilidade da barreira hematoencefálica é provavelmente de grande importância para a eliminação do vírus da raiva, e abordagens terapêuticas devem ser utilizadas para aumentar esta permeabilidade. Terapias que promovam o fechamento de barreiras hematoencefálicas, como a administração de corticosteroides, devem ser evitadas no manejo de pacientes humanos acometidos de raiva.
ABORDAGENS À TERAPIA DA RAIVA HUMANA
Não há uma terapia estabelecida para pacientes humanos com raiva. Uma abordagem ao tratamento da raiva humana foi recentemente publicada24. O artigo recomendou o uso de uma combinação de terapias e discutiu os prós e contras de se utilizar uma variedade de medicamentos específicos. Em 2004, uma paciente de 15 anos de idade, sem ter sido vacinada previamente ao aparecimento dos sintomas da doença clínica, sobreviveu à raiva50. Ela fora mordida no dedo por um morcego e não procurou atendimento médico ou recebeu qualquer tratamento àquela época. Por volta de um mês após a mordida, a paciente desenvolveu sintomas clínicos de raiva, e anticorpos neutralizantes contra o vírus da raiva foram detectados em seu soro e líquido cefalorraquidiano. Sua terapia incluiu coma terapêutico (induzido) com uso de midazolam intravenoso por sete dias; um padrão de supressão foi mantido em seu eletroencefalograma, e fenobarbital suplementar foi ministrado. Ela também recebeu terapia com ketamina e terapia antiviral, incluindo ribavirina e amantadina. Com sua melhora, a paciente recebeu alta do hospital apresentando déficit neurológico, porém apresentou melhora neurológica posteriormente11. Este caso foi o primeiro registro de sobrevivência à raiva sem que o indivíduo tenha sido vacinado antes do aparecimento dos sintomas da raiva clínica. Não se sabe se a terapia com um ou mais agentes específicos exerceu um papel importante para o resultado positivo do paciente16. Entretanto, daquele momento em diante, houve o registro de no mínimo 13 casos nos quais os principais componentes desta abordagem (o protocolo "Milwaukee") foram utilizados e que resultaram em desfechos fatais17. A indução ao coma per se não tem demonstrado ser útil para o tratamento de doenças infecciosas no sistema nervoso, e não há evidências que deem suporte a esta abordagem para o tratamento da raiva ou outras encefalites virais. Diante do exposto, o coma terapêutico não deve se tornar uma terapia de rotina para o tratamento da raiva.
O desenvolvimento precoce de anticorpos neutralizantes contra o vírus da raiva no curso clínico do paciente, que provavelmente ocorre em menos de 20% de todos os que são acometidos pela raiva, provavelmente contribuiu para um desfecho favorável. Os vírus da raiva de morcego podem ser menos neurovirulentos que as variantes caninas e outras responsáveis pela maioria dos casos humanos de raiva31, e a raiva causada pelas variantes caninas do vírus rábico pode levar a resultados piores que a causada por variantes de morcego. Finalmente, a maioria dos sobreviventes apresentou anticorpos neutralizantes contra o vírus da raiva em soros e líquido cefalorraquidiano, porém outros testes laboratoriais de diagnóstico normalmente apresentam resultado negativo para o antígeno do vírus da raiva e para o RNA em líquidos e tecidos (tecidos cerebrais não foram testados). Isso deve ocorrer devido à eficiente depuração viral.
CONCLUSÃO
As bases da disfunção neuronal na raiva são complexas, porém podem abranger mudanças degenerativas que envolvam processos neuronais, tais como dendritos e axônios. Por razões ainda não esclarecidas, a lesão neuronal é maior nos gânglios espinhais do que nos neurônios do SNC. A permeabilidade a imunoefetores da barreira hematoencefálica configurase importante para a depuração viral e a recuperação do paciente com raiva. Não há nenhuma terapia efetiva contra a raiva humana. Esperamos que uma melhor compreensão acerca da patogênese da doença leve ao desenvolvimento de novas terapias para a raiva humana.
REFERÊNCIAS
1 Allsopp TE, Fazakerley JK. Altruistic cell suicide and the specialized case of the virus-infected nervous system. Trends Neurosci. 2000 Jul;23(7):284-90. DOI:10.1016/S0166-2236(00)01591-5 [ Links ]
2 Bouzamondo E, Ladogana A, Tsiang H. Alteration of potassium-evoked 5-HT release from virus-infected rat cortical synaptosomes. Neuro Report. 1993 May;4(5):555-8. [ Links ]
3 Ceccaldi PE, Fillion MP, Ermine A, Tsiang H, Fillion G. Rabies virus selectively alters 5-HT1 receptor subtypes in rat brain. Eur J Pharmacol. 1993 Apr;245(2):129-38. [ Links ]
4 Charlton KM, Nadin-Davis S, Casey GA, Wandeler AI. The long incubation period in rabies: delayed progression of infection in muscle at the site of exposure. Acta Neuropathol. 1997 Jul;94(1):73-7. [ Links ]
5 Dhingra V, Li X, Liu Y, Fu ZF. Proteomic profiling reveals that rabies virus infection results in differential expression of host proteins involved in ion homeostasis and synaptic physiology in the central nervous system. J Neurovirol. 2007 Apr;13(2):107-17. [ Links ]
6 Dumrongphol H, Srikiatkhachorn A, Hemachudha T, Kotchabhakdi N, Govitrapong P. Alteration of muscarinic acetylcholine receptors in rabies viral-infected dog brains. J Neurol Sci. 1996 Apr;137(1):1-6. DOI:10.1016/0022-510X(95)00275-7 [ Links ]
7 Fazakerley JK, Allsopp TE. Programmed cell death in virus infections of the nervous system. Curr Top Microbiol Immunol. 2001;253:95-119.
8 Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000 Oct;28(1):41-51. [ Links ]
9 Griffin DE, Hardwick JM. Perspective: virus infections and the death of neurons. Trends Microbiol. 1999 Apr;7(4):155-60. DOI:10.1016/S0966-842X(99)01470-5 [ Links ]
10 Hooper DC, Ohnishi ST, Kean R, Numagami Y, Dietzschold B, Koprowski H. Local nitric oxide production in viral and autoimmune diseases of the central nervous system. Proc Natl Acad Sci U S A. 1995 Jun;92(12):5312-6. [ Links ]
11 Hu WT, Willoughby RE Jr, Dhonau H, Mack KJ. Longterm follow-up after treatment of rabies by induction of coma (Letter). N Engl J Med. 2007 Aug;357(9):945-6.
12 Iwata M, Komori S, Unno T, Minamoto N, Ohashi H. Modification of membrane currents in mouse neuroblastoma cells following infection with rabies virus. Br J Pharmacol. 1999 Apr;126(8):1691-8. DOI:10.1038/sj.bjp.0702473 [ Links ]
13 Jackson AC. Cholinergic system in experimental rabies in mice. Acta Virol. 1993 Dec;37(6):502-8. [ Links ]
14 Jackson AC. Human disease. In: Jackson AC, Wunner WH, editors. Rabies. 2nd ed. London: Elsevier Academic Press; 2007. p. 309-40.
15 Jackson AC. Pathogenesis. In: Jackson AC, Wunner WH, editors. Rabies, 2nd ed. London: Elsevier Academic Press; 2007. p. 341-81.
16 Jackson AC. Recovery from rabies. N Engl J Med. 2005 Jun;352(24):2549-50.
17 Jackson AC. Update on rabies diagnosis and treatment. Curr Infect Dis Rep. 2009 Jul;11(4):296- 301. [ Links ]
18 Jackson AC, Fenton MB. Human rabies and bat bites. Lancet. 2001 May;357(9269):1714.
19 Jackson AC, Park H. Apoptotic cell death in experimental rabies in suckling mice. Acta Neuropathol. 1998 Feb;95(2):159-64. [ Links ]
20 Jackson AC, Randle E, Lawrance G, Rossiter JP. Neuronal apoptosis does not play an important role in human rabies encephalitis. J Neurovirol. 2008 Oct;14(5):368-75. DOI:10.1080/13550280802216502 [ Links ]
21 Jackson AC, Rasalingam P, Weli SC. Comparative pathogenesis of recombinant rabies vaccine strain SAD-L16 and SAD-D29 with replacement of Arg333 in the glycoprotein after peripheral inoculation of neonatal mice: less neurovirulent strain is a stronger inducer of neuronal apoptosis. Acta Neuropathol. 2006 Apr;111(4):372-8. DOI:10.1007/s00401-005-0006-z [ Links ]
22 Jackson AC, Reimer DL. Pathogenesis of experimental rabies in mice: an immunohistochemical study. Acta Neuropathol. 1989;78(2):159-65. [ Links ]
23 Jackson AC, Rossiter JP. Apoptosis plays an important role in experimental rabies virus infection. J Virol. 1997 Jul;71(7):5603-7. [ Links ]
24 Jackson AC, Warrell MJ, Rupprecht CE, Ertl HC, Dietzschold B, O'Reilly M, et al. Management of rabies in humans. Clin Infect Dis. 2003 Jan;36(1):60-3. [ Links ]
25 Jackson AC, Wunner WH. Rabies. 2nd ed. London: Elsevier Academic Press; 2007.
26 Juntrakul S, Ruangvejvorachai P, Shuangshoti S, Wacharapluesadee S, Hemachudha T. Mechanisms of escape phenomenon of spinal cord and brainstem in human rabies. BMC Infect Dis. 2005 Nov;5(1):104.
27 Kelly RM, Strick PL. Rabies as a transneuronal tracer of circuits in the central nervous system. J Neurosci Methods. 2000 Nov;103(1):63-71. DOI:10.1016/S0165-0270(00)00296-X [ Links ]
28 Koprowski H, Zheng YM, Heber-Katz E, Fraser N, Rorke L, Fu ZF, et al. In vivo expression of inducible nitric oxide synthase in experimentally induced neurologic diseases. Proc Natl Acad Sci U S A. 1993 Apr;90(7):3024-7. Erratum in: Proc Natl Acad Sci U S A. 1993 Jun;90(11):5378. [ Links ]
29 Kucera P, Dolivo M, Coulon P, Flamand A. Pathways of the early propagation of virulent and avirulent rabies strains from the eye to the brain. J Virol. 1985 Jul;55(1):158-62. [ Links ]
30 Ladogana A, Bouzamondo E, Pocchiari M, Tsiang H. Modification of tritiated γ-amino-η-butyric acid transport in rabies virus-infected primary cortical cultures. J Gen Virol. 1994 Mar;75(Pt 3):623-7. [ Links ]
31 Lafon M. Bat rabies: the Achilles heel of a viral killer? Lancet. 2005 Sep;366(9489):876-7.
32 Lentz TL, Burrage TG, Smith AL, Crick J, Tignor GH. Is the acetylcholine receptor a rabies virus receptor? Science. 1982 Jan;215(4529):182-4. [ Links ]
33 Li XQ, Sarmento L, Fu ZF. Degeneration of neuronal processes after infection with pathogenic, but not attenuated, rabies viruses. J Virol. 2005 Aug;79(15):10063-8. DOI:10.1128/JVI.79.15.10063–10068.2005 [ Links ]
34 Lycke E, Tsiang H. Rabies virus infection of cultured rat sensory neurons. J Virol. 1987 Sep;61(9):2733-41. [ Links ]
35 Morimoto K, Hooper DC, Spitsin S, Koprowski H, Dietzschold B. Pathogenicity of different rabies virus variants inversely correlates with apoptosis and rabies virus glycoprotein expression in infected primary neuron cultures. J Virol. 1999 Jan;73(1):510-8. [ Links ]
36 Morimoto K, Patel M, Corisdeo S, Hooper DC, Fu ZF, Rupprecht CE, et al. Characterization of a unique variant of bat rabies virus responsible for newly emerging human cases in North America. Proc Natl Acad Sci U S A. 1996 May;93(11):5653-8. [ Links ]
37 Noah DL, Drenzek CL, Smith JS, Krebs JW, Orciari L, Shaddock J, et al. Epidemiology of human rabies in the United States, 1980 to 1996. Ann Intern Med. 1998 Jun;128(11):922-30. [ Links ]
38 Prosniak M, Hooper DC, Dietzschold B, Koprowski H. Effect of rabies virus infection on gene expression in mouse brain. Proc Natl Acad Sci U S A. 2001 Feb;98(5):2758-63. DOI: 10.1073/pnas.051630298 [ Links ]
39 Reid JE, Jackson AC. Experimental rabies virus infection in Artibeus jamaicensis bats with CVS-24 variants. J Neurovirol. 2001 Dec;7(6):511-7. DOI: 10.1080/135502801753248097 [ Links ]
40 Rossiter JP, Hsu L, Jackson AC. Selective vulnerability of dorsal root ganglia neurons in experimental rabies after peripheral inoculation of CVS-11 in adult mice. Acta Neuropathol. 2009 Aug;118(2):249-59. [ Links ]
41 Roy A, Hooper DC. Lethal silver-haired bat rabies virus infection can be prevented by opening the blood-brain barrier. J Virol. 2007 Aug;81(15):7993-8. DOI:10.1128/JVI.00710-07 [ Links ]
42 Roy A, Phares TW, Koprowski H, Hooper DC. Failure to open the blood-brain barrier and deliver immune effectors to central nervous system tissues leads to the lethal outcome of silver-haired bat rabies virus infection. J Virol. 2007 Feb;81(3):1110-8. DOI: 10.1128/JVI.01964-06 [ Links ]
43 Scott CA, Rossiter JP, Andrew RD, Jackson AC. Structural abnormalities in neurons are sufficient to explain the clinical disease and fatal outcome of experimental rabies in yellow fluorescent protein-expressing transgenic mice. J Virol. 2008 Jan;82(1):513-21. DOI:10.1128/JVI.01677-07 [ Links ]
44 Smith JS, Fishbein DB, Rupprecht CE, Clark K. Unexplained rabies in three immigrants in the United States. A virologic investigation. N Engl J Med. 1991 Jan;324(4):205-11. [ Links ]
45 Tang Y, Rampin O, Giuliano F, Ugolini G. Spinal and brain circuits to motoneurons of the bulbospongiosus muscle: retrograde transneuronal tracing with rabies virus. J Comp Neurol. 1999 Nov;414(2):167-92. [ Links ]
46 Theerasurakarn S, Ubol S. Apoptosis induction in brain during the fixed strain of rabies virus infection correlates with onset and severity of illness. J Neurovirol. 1998 Aug;4(4):407-14. [ Links ]
47 Tsiang H, Ceccaldi PE, Lycke E. Rabies virus infection and transport in human sensory dorsal root ganglia neurons. J Gen Virol. 1991 May;72(Pt 5):1191-4.
48 Tsiang H. Neuronal function impairment in rabies-infected rat brain. J Gen Virol. 1982 Aug;61(Pt 2):277-81. [ Links ]
49 Weli SC, Scott CA, Ward CA, Jackson AC. Rabies virus infection of primary neuronal cultures and adult mice: failure to demonstrate evidence of excitotoxicity. J Virol. 2006 Oct;80(20):10270-3. DOI:10.1128/JVI.01272-06 [ Links ]
50 Willoughby RE Jr, Tieves KS, Hoffman GM, Ghanayem NS, Amlie-Lefond CM, Schwabe MJ, et al. Survival after treatment of rabies with induction of coma. N Engl J Med. 2005 Jun;352(24):2508-14. [ Links ]
51 World Health Organization. WHO expert consultation on rabies: first report. Geneva; 2005. p. 1-88.
 Correspondência/Correspondence/Correspondencia:
Correspondência/Correspondence/Correspondencia:
Alan C. Jackson
Health Sciences Centre
GF-543, 820 Sherbrook Street
Winnipeg, Manitoba R3A 1R9 Canada
Phone: 204-787-1578 Fax: 204-787-1486
E-mail: ajackson2@hsc.mb.ca
Recebido em/Received/Recibido en: 21/06/2009
Aceito em/Accepted/Aceito en: 02/10/2009