








Servicios Personalizados
Revista

Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Pan-Amazônica de Saúde
versión impresa ISSN 2176-6215versión On-line ISSN 2176-6223
Rev Pan-Amaz Saude v.1 n.3 Ananindeua sep. 2010
http://dx.doi.org/10.5123/S2176-62232010000300018
RELATO DE CASO | CASE REPORT | RELATO DE CASO
Síndrome Dubin-Johnson: importante causa de icterícia colestática na infância
Dubin-Johnson syndrome: an important cause of obstructive jaundice in children
El síndrome de Dubin-Johnson: importante causa de ictericia colestática en la infancia
Maria Cleonice Aguiar JustinoI; Eliana Canen Pinto SoaresII; Cláudio Sérgio Carvalho de AmorimI
IInstituto de Ciências da Saúde, Universidade Federal do Pará, Belém, Pará, Brasil
IIFundação Santa Casa de Misericórdia do Pará, Belém, Pará, Brasil
Endereço para correspondência
Correspondence
Dirección para correspondencia
RESUMO
A síndrome Dubin-Johnson é caracterizada clinicamente por episódios de icterícia colestática recorrente, de caráter benigno e familiar, sendo definida pela presença de pigmento melânico nos hepatócitos. Os autores relatam um caso de síndrome Dubin-Johnson em uma criança portadora de icterícia e hepatoesplenomegalia, cujo diagnóstico foi confirmado pela presença do pigmento castanho-escuro à microscopia hepática realizada a partir de biópsia, e alertam para a necessidade de suspeição dessa síndrome em casos de icterícia colestática flutuante na infância.
Palavras-chave: Colestase; Icterícia; Icterícia Idiopática Crônica.
ABSTRACT
The Dubin-Johnson syndrome is clinically characterized by recurrent episodes of benign and familial obstructive jaundice. It is identified by the presence of melanic pigment in the hepatocytes. The authors report a case of Dubin-Johnson syndrome in a child with jaundice and hepatosplenomegaly, whose diagnosis was confirmed by the presence of dark brown pigment on microscopy of liver biopsy. They suggest the suspicion of this syndrome in cases of fluctuating obstructive jaundice in children.
Keywords: Cholestasis; Jaundice; Jaundice, Chronic Idiopathic.
RESUMEN
El síndrome Dubin-Johnson se caracteriza clínicamente por episodios de ictericia colestática recurrente, de carácter benigno y familiar, siendo definido por la presencia de pigmento melánico en los hepatocitos. Los autores relatan un caso de síndrome Dubin-Johnson en una nina portadora de ictericia y hepatoesplenomegalia, diagnóstico que fue confirmado por la presencia del pigmento castano oscuro en la microscopia hepática realizada a partir de biopsia, y alertan para la necesidad de sospecha de ese síndrome en casos de ictericia colestática fluctuante en la infancia.
Palabras clave: Cholestasis; Ictericia; Ictericia Idiopática Crónica.
INTRODUÇÃO
Em 1954, foram publicadas as primeiras descrições de icterícia benigna e familiar no curso da qual ocorre hiperbilirrubinemia plasmática às custas da fração direta, caracterizada pelo depósito nas células hepáticas de pigmento melânico característico1. A síndrome Dubin-Johnson é uma patologia rara, geneticamente determinada, de herança autossômica recessiva, favorecida, portanto, pela consanguinidade. A icterícia evolui em surtos, frequentemente precipitados por cansaço, fortes emoções, exercícios físicos ou infecções intercorrentes, acompanhada de hepatomegalia discreta e colúria2,3,4.
Nesta síndrome, a icterícia inicia na infância, por vol ta dos 2 anos de idade, podendo surgir, excepcionalmente, ainda no período neonatal4. A instalação do quadro pode acontecer de forma aguda e acompanhada de febre, semelhante a uma hepatite viral5,3. Não há sinais hematológicos atribuídos a hemólises; a hiperbilirrubinemia ocorre de forma intermitente, com predomínio da fração direta, e as diferentes provas de função hepática apresentam valores normais5,3. Na puberdade, o quadro clínico apresenta-se com icterícia em 100% dos casos, dor abdominal, especialmente em hipocôndrio direito, hepatomegalia, fraqueza, anorexia, coluria e hipocolia fecal1. Alguns pacientes apresentam alterações na excreção urinária das coproporfirinas, como diminuição da coproporfirina III e aumento relativo do isômero I, podendo estas alterações serem encontradas em graus discretos em familiares6.
A síndrome ocorre devido à expressão defeituosa do gene MRP2, um transportador de membrana canalicular dependente de ATP7,8,9. O diagnóstico é estabelecido a partir da realização de colecistografia oral, teste da bromossulfaleina e biopsia hepática, associados ao quadro clínico5,3,4.
A biópsia hepática é o exame padrão ouro para o diagnóstico da síndrome, evidenciando a presença de pigmento escuro granuloso nos lissossomos dos hepatócitos centrolobulares10,11.
Na literatura, a maioria dos casos descritos envolve adultos jovens1,5,4,11, e, nos relatos envolvendo crianças, normalmente há história pregressa de icterícia colestática de remissão espontânea no período neonatal sem diagnóstico estabelecido2,12,3,13,9,14.
RELATO DE CASO
ANAMNESE
A.M.A., sexo feminino, 2 anos e 8 meses de idade, cor parda, natural e procedente do Município de Augusto Corrêa, nordeste do Estado do Pará. A paciente foi admitida em enfermaria de pediatria, doente há oito meses, com quadro clínico de distensão abdominal e episódios de febre intermitente, associados à palidez cutâneo-mucosa e icterícia flutuante. A criança é filha de pais consanguíneos e tem história de ter apresentado três episódios de malária nos últimos oito meses, tratados no Município de sua procedência. Não há relato prévio de icterícia.
EXAME FÍSICO
Presença de icterícia, palidez cutâneo-mucosa e desnutrição. Abdome distendido, normotenso, doloroso à palpação superficial e profunda no quadrante superior direito, com fígado palpável a 7 cm do rebordo costal direito e baço palpável a 11 cm do rebordo costal esquerdo.
EXAMES COMPLEMENTARES
Bilirrubina total de 3,2 mg/dL, com bilirrubina direta de 2,5 mg/dL. Eletroforese de hemoglobina e de proteínas dentro da normalidade. Sorologia não reagente para hepatites virais, toxoplasmose, sífilis, rubéola, herpes, citomegalovírus, HIV e calazar. Pesquisa de plasmódio negativa. Mielograma sem alterações.
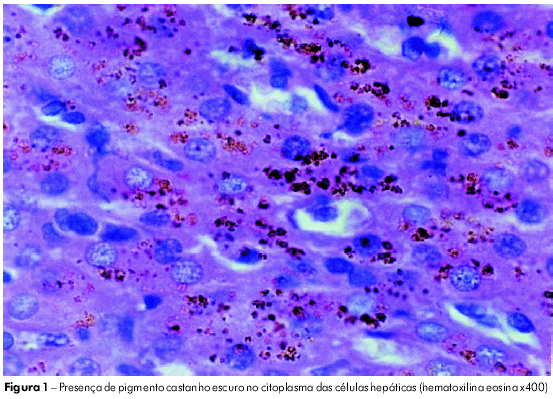
A ultrassonografia abdominal revelou hepatoesplenomegalia com ecotextura homogênea, sem outras alterações. A biópsia hepática à microscopia ótica revelou arquitetura do órgão preservada e hepatócitos com depósitos citoplasmáticos de pigmento granular fino, de coloração castanho-escuro, distribuídos por todo o lóbulo, sendo Perls negativo e PAS positivo, compatível com síndrome Dubin-Johnson (Figura 1). Não foram realizados os exames de colecistrografia oral, teste da bromosulfaleina e pesquisa da mutação pela biologia molecular, por dificuldades técnicas.
EVOLUÇÃO
A paciente evoluiu com resolução espontânea da icterícia, ocorrendo regressão lenta da hepatoesplenomegalia. Após 25 dias de internação, obteve alta hospitalar, sendo referenciada para acompanhamento ambulatorial.
DISCUSSÃO
A síndrome Dubin-Johnson é uma doença rara, de transmissão genética, cuja base molecular é um defeito no gene que codifica a proteína transportadora de ânions orgânicos MRP28. Apesar da natureza genética, há casos descritos na literatura sem história familiar da doença6. No presente relato, a paciente, filha de pais consanguíneos, não referia qualquer história familiar de sintomas compatíveis com a síndrome.
O início dos sintomas, aos 2 anos e 8 meses de idade, foi compatível com a faixa etária descrita pela literatura em relação ao surgimento da icterícia. É provável que as infecções por Plasmodium, ocasionando malária, tenham contribuído para a manifestação da síndrome Dubin-Johnson pela própria hemólise significativa que essa infecção apresenta.
A hepatomegalia discreta é um dos sintomas presentes na síndrome, não havendo relatos de esplenomegalia associada. A acentuada hepatoesplenomegalia encontrada na paciente pode ser atribuída aos sucessivos episódios de malária ocorridos nos meses que antecederam sua hospitalização, uma vez que tal envolvimento faz parte do quadro dessa doença na infância15.
CONSIDERAÇÕES FINAIS
Embora a síndrome Dubin-Johnson seja uma patologia rara, é de extrema relevância incluí-la na investigação diagnóstica de casos de icterícia flutuante na infância. O prognóstico é favorável e sua evolução benigna, não sendo requerido qualquer tratamento. Portanto, estabelecer corretamente o diagnóstico torna-se importante para prevenir procedimentos futuros desnecessários. Às portadoras da síndrome, por ocasião da idade adulta, apenas orienta-se não fazer uso de contraceptivos orais, os quais podem competir com a secreção hepatocitária de ânions orgânicos.
REFERÊNCIAS
1 Dubin IN, Johnson FB. Chronic idiopathic jaundice with unidentifield pigment in liver cells: a new clinicopathologic entity with a report of 12 cases. Medicine (Baltimore). 1954 Sep;33(3):155-97.
2 Haimi-Cohen Y, Amir J, Merlob P. Neonatal and infantile Dubin-Johnson syndrome. Pediatr Radiol. 1998 Nov;28(11):900. DOI:10.1007/s002470050494 [ Links ]
3 Kondo T, Yagy R, Kuchiba K. Dubin-Johnson syndrome in neonate. N Engl J Med. 1975;292:1028-9.
4 Rastogi A, Krishnani N, Pandey R. Dubin-Johnson syndrome, a clinicopathologic study of twenty cases. Indian J Pathol Microbiol. 2006;49(4):500-4. [ Links ]
5 Dubin IN. Chronic idiopathic jaundice with: a review of fifty cases. Am J Med. 1958 Feb;24(2):268-92.
6 Lanosa RA, Mazzini O, Pietriangelo C, Celia EJ, Monserrat JM. Contribucion al diagnostico del síndrome de Dubin-Johnson. Acta Gastroenterol Latinoam.1980;10:1-12.
7 Cebecauerova D, Jirasek T, Budisova L, Mandys V, Volf U, Novotna Z, et al. Dual hereditary jaundice: simultaneous occurrence of mutations causing Gilbert's and Dubin-Johnson syndrome. Gastroenterology. 2005 Jul;129(1):135-320. [ Links ]
8 Paulusma CC, Kool M, Bosma PJ, Scheffer GL, Borg F, Sheper RJ, et al. A mutation in the human canalicularmultispecific organic anion transporter gene causes the Dubin-Johnson syndrome. Hepatology. 1997;25(6):1539-42.
9 Stapelbroek JM, Van Erpecum KJ, Klomp LWJ, Houwen RHJ. Liver disease associated with canalicular transport defects: current and future therapies. Journal of Hepatology. 2010 Feb;52(2):258-71. [ Links ]
10 Baba N, Ruppert RD. The Dubin-Johnson syndrome: electron microscopic observation of hepatic pigment-a case study. Am J Clin Pathol. 1972 Mar;57(3):306-10.
11 Sobaniec-Lotwska ME, Lebensztejn DM. Ultrastructure of Kupffer cells and hepatocytes in the Dubin-Johnson syndrome: a case report. World J Gastroenterol. 2006 Feb;12(6):987-9.
12 Kimura A, Ushijima K, Kage M, Mahara R, Tohma M, Inokuchi T, et al. Neonatal Dubin-Johnson syndrome with severe cholestasis: effective phenobarbital therapy. Acta Paediatr Scand. 1991 Mar;80(3):381-5. [ Links ]
13 Shieh CC, Chang MH, Chen CL. Dubin-Jhonson syndrome presenting with neonatal cholestasis. Arch Dis Child. 1990;65:898-9. Doi:10.1136/Adc.65.8.898 [ Links ]
14 Tsai WH, Teng RJ, Chu JS, Chang MH, Ho MM. Neonatal Dubin- Johnson syndrome. J Pediatr Gastroenterol Nutr. 1994;18(2):253-4.
15 Amaral CN, Albuquerque YD, Pinto AYN, Souza JM. A importância do perfil clínico-laboratorial no diagnóstico diferencial entre malária e hepatite aguda viral. J Pediatr (Rio J). 2003;79(5):429-34. [ Links ]
 Correspondência / Correspondence / Correspondencia:
Correspondência / Correspondence / Correspondencia:
Serviço de Pediatria,
Instituto de Ciências da Saúde,
Universidade Federal do Pará
Praça Camilo Salgado, 01.
Bairro: Umarizal
CEP:66050-060
Belém-Pará-Brasil
E-mail:mariajustino@iec.pa.gov.br
Recebido em / Received / Recibido en: 29/7/2010
Aceito em / Accepted / Aceito en: 28/9/2010