








Serviços Personalizados
Journal

Artigo
 texto em
texto em  Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Pan-Amazônica de Saúde
versão impressa ISSN 2176-6215versão On-line ISSN 2176-6223
Rev Pan-Amaz Saude v.1 n.4 Ananindeua dez. 2010
http://dx.doi.org/10.5123/S2176-62232010000400013
ARTÍCULO DE REVISIÓN
La contribución de los polimorfismos humanos del eritrocito en la protección contra la malaria
Patrícia Machado*; Cristina Mendes*; Virgílio Estólio do Rosário; Ana Paula Arez
Unidade de Ensino e Investigação de Malária, Centro de Malária e outras Doenças Tropicais, Laboratório Associado, Instituto de Higiene e Medicina Tropical, Lisboa, Portugal
Endereço para correspondência
Correspondence
Dirección para correspondencia
Título original: A contribuição dos polimorfismos humanos do eritrócito na proteção contra a malária. Traducido por: Lota Moncada
RESUMEN
La comprensión del complejo ciclo de vida de la malaria ha aumentado mucho en los últimos años pero, a pesar de décadas de investigación y lucha contra esa enfermedad, esta continúa a ser uno de los principales problemas de salud pública, especialmente en las áreas más pobres del planeta. Debido a su elevada prevalencia en ciertas regiones del globo, desde hace cerca de 10 mil años, la malaria ha ejercido una presión selectiva muy fuerte sobre el genoma humano. El componente genético de susceptibilidad al parásito es complejo, con una variedad de polimorfismos influyendo en la patogénesis y la respuesta del hospedero, y uno de los desafíos en la lucha contra esta enfermedad es evaluar estos determinantes de susceptibilidad y descifrar los mecanismos involucrados para utilizarlos como nuevas metas para fármacos o vacunas. Entre los polimorfismos genéticos humanos descritos como protectores contra la malaria, lo más comunes y mejor caracterizados involucran a proteínas estructurales específicas (tales como las hemoglobinas S y C, las talasemias, el antígeno Duffy y el grupo sanguíneo O) y enzimas eritrocitarias (como la deficiencia de glucosa-6-fosfato deshidrogenasa, y, más recientemente descrita, la deficiencia de piruvato quinasa). Esta pequeña revisión aborda estas variantes genéticas y discute algunos de los resultados controvertidos obtenidos, así como los mecanismos que pueden justificar esta protección.
Palabras clave: Malaria; Polimorfismo Genético; Anemia Hemolítica Congénita; Hemoglobinas Anormales; Sistema del Grupo Sanguíneo Duffy; Sistema del Grupo Sanguíneo ABO.
INTRODUCCIÓN
Según la Organización Mundial de Salud, la malaria continúa a ser una de las principales causas de morbilidad y mortalidad en los países tropicales. Se estima que apenas en el año de 2008 haya causado cerca de 243 millones de casos clínicos que resultaron en, aproximadamente, 863 mil muertes1. La malaria es una enfermedad infecciosa, causada por parásitos protozoarios del género Plasmodium y transmitida a los humanos por picadas de mosquitos infectados del género Anopheles. Las cinco especies de parásitos que infectan a los humanos son: Plasmodium falciparum, Plasmodium ovale, Plasmodium malariae, Plasmodium vivax y Plasmodium knowlesi. Las especies P. vivax y P. falciparum son las más comunes, siendo esta última responsable por las formas graves de la enfermedad-malaria cerebral o anemia grave.
Siendo una enfermedad de muy alta prevalencia desde hace miles de años, la malaria ha ejercido una presión selectiva sobre el genoma humano2,3, especialmente en los eritrocitos, que desempeñan un papel fundamental, como células huéspedes, en el ciclo de vida del parásito. Así, los genes que afectan a la estructura y/o a la funcionalidad de los eritrocitos son los que presentan mayor número de variantes genéticas descritas como asociadas a la protección contra la malaria o sus síntomas4, como es el caso de algunas hemoglobinopatías, las talasemias, el antígeno Duffy, el sistema ABO, la deficiencia de glucosa-6-fosfato deshidrogenasa (G6PD) y, más recientemente estudiado, el déficit de piruvato quinasa (PK).
En los últimos años han surgido nuevas líneas de investigación de la malaria, como las que se vuelcan sobre la estrecha relación entre el huésped y el parásito, que explica, por un lado, la elevada abundancia de P. falciparum en África, y por otro, el motivo por el que la mayoría de los individuos parasitados no desarrollen complicaciones de malaria severa, mientras otros sucumban a la enfermedad5.
En esta pequeña revisión se abordan algunos de los polimorfismos del eritrocito humano, de los más tradicionales a los más recientemente identificados, descritos en trabajos anteriores como protectores contra la malaria, y se discuten los resultados controvertidos que se ha obtenido a lo largo del tiempo, bien como algunos de los posibles mecanismos subyacentes a esta protección. Se da especial énfasis a las especies de parásitos P. falciparum y P. vivax por ser las especies más comunes que infectan a los humanos, siendo P. falciparum la más mortal, y también se da especial atención a regiones del África subsahariana, por ser las más afectadas.
LA SUSCEPTIBILIDAD A LA MALARIA ES UNA CARACTERÍSTICA HEREDITARIA
La mayoría de las picadas de mosquitos infectados no produce enfermedad (infección con sintomatología clásica de fiebre, dolores de cabeza, entre otros). Los niños expuestos a tales picadas o no son infectados (cerca de la mitad), o tienen infección sin síntomas (cerca de 25%), o tienen infección con fiebre y otros síntomas sin gravedad (aproximadamente 25%). Apenas en raros casos los niños desarrollan manifestaciones de malaria severa como la anemia grave, el coma y la malaria cerebral. Greenwood y colaboradores6 estimaron que en áreas de elevada transmisión, en 400 picadas potencialmente infecciosas, apenas 200 resultan en infección y solamente dos originan malaria grave y una, la muerte.
Los factores que determinan el desarrollo de la infección y la enfermedad son pues, importantes, y deben ser clarificados. Podemos hablar de la combinación de diversos aspectos interconectados: la tasa de inoculación del mosquito, la dosis de esporozoítos, la inmunidad adquirida de infecciones anteriores, la virulencia del parásito, los polimorfismos genéticos del huésped humano, el estado de nutrición del individuo infectado, las condiciones ambientales y el acceso a un tratamiento eficiente. La prevalencia de la infección de P. falciparum es, a pesar de todo, suficientemente alta para matar a 1 millón de personas en África por año, donde la tasa de mortalidad en niños menores de 5 años llega a ser entre 1 y 2%1,7,8.
Los factores genéticos del huésped humano dan una contribución significativa a la diversidad observada en la malaria. En una misma población, existe un elevado grado de variación entre individuos con relación a los fenotipos de susceptibilidad a la malaria, incluyendo la carga parasitaria, la incidencia de la enfermedad y la severidad6 y la magnitud y tipo de respuesta inmune a los antígenos de la malaria9,10.
Cuando la base genética de algunos desórdenes del eritrocito fue inicialmente investigada, los científicos se depararon con la extraña paradoja de la presencia de elevadas frecuencias de mutaciones deletéreas en algunas poblaciones. La talasemia, por ejemplo, que es la base de una anemia microcítica, es muy frecuente en varias regiones del Mediterráneo, Oriente Medio, África y Suroeste de Asia. Haldane11, en 1949, propuso que un alelo mutado alcanza y mantiene una elevada frecuencia, no por medio de una tasa excepcionalmente alta de mutación, y sí porque es consecuencia de una ventaja selectiva contra la malaria causada por P. falciparum, cuya distribución coincide con la de la talasemia.
Para evaluar el impacto de la determinación genética de la susceptibilidad a la malaria clínica, se hizo un estudio longitudinal con niños mellizos en Gambia12 en el que se observó que existe una mayor probabilidad de ambos gemelos monozigóticos de desarrollar fiebre provocada por la malaria que en gemelos dizigóticos, lo que indica que hay factores genéticos involucrados en el desarrollo de la enfermedad. En otro estudio desarrollado en Sri-Lanka13, se estimó que el factor hereditario es el responsable por cerca de 15% de la incidencia de infección sintomática y asintomática por P. falciparum y aproximadamente por 10% de la intensidad de los síntomas clínicos. En un estudio en Kenia14, fue monitoreada, en dos cohortes de niños, la incidencia de malaria clínica no complicada y las admisiones hospitalarias debido a la malaria. En ambos casos, se estimó que 25% de la variación total era explicada por la suma de los efectos de los genes del huésped y, de estos, la hemoglobina S, el factor genético de resistencia a la malaria más conocido, explicaba apenas 2% de la variación total, sugiriendo la existencia de muchos genes protectores desconocidos, cada uno resultando en pequeños efectos en las poblaciones.
LOS DESÓRDENES DE LA HEMOGLOBINA
La hemoglobina normal de un adulto se compone por dos cadenas de α-globina y dos de β-globina. Los desórdenes de la hemoglobina pueden ser divididos en dos grupos: uno en el que hay una disminución de la producción de las formas estructurales normales de las α- o β-globinas, dando origen a las α- o β-talasemias, respectivamente; y otra en la que hay producción de formas estructurales mutantes (hemoglobinopatías), como en el caso de las hemoglobinas S (HbS), C (HbC) o E (HbE). A pesar de que fueron identificadas centenas de variantes estructurales de la hemoglobina, apenas las tres referidas anteriormente presentan frecuencias polimórficas. Las dos primeras son muy frecuentes especialmente en África Occidental15 y la hemoglobina E es común en el Sudeste Asiático16.
Las cadenas de la β-globina son codificadas por un único gen que se localiza en el cromosoma 11; las cadenas a son codificadas por dos genes íntimamente conectados en el cromosoma 16. Así, en un individuo normal diploide, existen dos loci que codifican para la cadena β y cuatro que codifican para la cadena α.
LA DREPANOCITOSIS O ANEMIA FALCIFORME
La drepanocitosis o anemia falciforme es una hemoglobinopatía de carácter genético, causada por una mutación en la posición 6 del gen de la β-globina (HbB), ocurriendo la sustitución del ácido glutámico por valina (β6Glu>Val). La forma anormal producida, denominada hemoglobina S, hace con que los glóbulos rojos se vuelvan rígidos y con forma de hoz, impidiendo que los mismos atraviesen los vasos sanguíneos y lleguen a los órganos19. Los efectos clínicos provocados por esta enfermedad son bastante variables, dependiendo de que los individuos sean portadores de rasgo drepanocítico (HbAS, que presenta el alelo normal A y el alelo mutado S) o si son individuos enfermos (HbSS, individuos homocigóticos para el alelo mutado S). Mientras que en los individuos portadores de síntomas de la enfermedad, cuando existen, son muy leves, los individuos enfermos presentan síntomas graves como anemias severas, infecciones graves y lesiones en órganos vitales, provocando una disminución en la esperanza media de vida4. La variante S de la hemoglobina se halla ampliamente distribuida por todo el mundo, presentando frecuencias muy elevadas en el África subsahariana (especialmente en la región occidental), en el Oriente Medio y en algunas zonas de India15.
La drepanocitosis fue una de las primeras hemoglobinopatías a ser asociadas a una protección contra la malaria. Anthony Allison20, en 1954, constató que los eritrocitos de los individuos con rasgo drepanocítico eran más difícilmente parasitados por P. falciparum que los normales, concluyendo que los individuos heterocigóticos tendrían una ventaja selectiva en las regiones hiperendémicas de malaria. Varias evidencias sugieren la existencia de un equilibrio entre la eliminación del alelo mutado (alelo S) por muerte precoz de los homocigóticos y su preservación en los heterocigóticos debido a esta ventaja selectiva. Los individuos portadores del alelo S parecen estar favorecidos en relación a los individuos no portadores, habiendo de esta forma la selección de este alelo, permitiendo que el mismo sea encontrado en un porcentaje de 5 a 40% en la población mundial15,21,22.
Varios estudios han sido realizados a lo largo de los últimos 60 años para comprender cual es el nivel de protección conferido por el estado heterocigótico. Aidoo et al23, Carter y Mendis24, Ayi et al25 y Williams et al26 demostraron que el genotipo HbAS está significativamente asociado a la protección contra la malaria severa provocada por el parásito P. falciparum. Aidoo et al23 demostraron que había una protección significativa del 60% en la mortalidad en niños entre los 2 y los 16 meses de edad. En niños con edades inferiores a 2 meses esa reducción no fue observada. Williams et al26 demostraron que había una protección de 90% en individuos heterocigóticos (HbAS) en casos de malaria severa y una protección de 50% en los casos de malaria asintomática.
Existe controversia sobre los mecanismos subyacentes a esta protección, pero algunos estudios la asocian a la dificultad que los parásitos tienen para invadir y crecer dentro de los eritrocitos con los genes mutados27. Sin embargo, otros estudios, como el de Cappadoro et al28, dicen no haber encontrado ninguna diferencia en el crecimiento de los parásitos en eritrocitos normales y en eritrocitos mutados. Otra explicación fue dada por Luzzato et al (1970) y Roth et al (1978) (in Williams et al29). Ellos verificaron que los eritrocitos HbAS parasitados tienen tendencia a quedar con una forma irregular más rápidamente que los no parasitados, llevando a la muerte intracelular del parásito. Un estudio más reciente30 mostró que la adhesión de eritrocitos parasitados AS a las células endoteliales microvasculares y a los monocitos es significativamente menor que la de los eritrocitos parasitados AA. Esta reducción está relacionada con alteraciones en lo principal, conectando citoadhesión y factor de virulencia del parásito (P. falciparum erythrocyte membrane protein-1, PfEMP-1).
En otro estudio31, se observó que el porcentaje de trofozoitos en la sangre periférica en niños asintomáticos con rasgo drepanocítico (HbAS) era de 75%, mientras que en los niños asintomáticos normales (HbAA) era de 37,5%, sugiriendo que hay un secuestro reducido de los eritrocitos parasitados en los niños HbAS. En los individuos HbAS pueden ocurrir alteraciones en la expresión de los ligandos de los eritrocitos infectados (como el PfEMP-1), reduciendo la citoadhesión y originando una infección menos grave. Estos resultados contrarían los obtenidos en el estudio anterior30.
LA HEMOGLOBINA C (HBC)
La hemoglobina C (HbC), tal como la hemoglobina S, es provocada por una mutación en la posición 6 del gen de la β-globina habiendo, en este caso, la sustitución del ácido glutámico por una lisina (β6Glu>Lys). La hemoglobina C se encuentra principalmente en África Occidental, aunque se a menos frecuente que la hemoglobina S, y resulta en un fenotipo menos severo que la drepanocitosis: los homocigóticos generalmente tienen una anemia hemolítica leve y los heterocigóticos no presentan una reducción significativa en los niveles de la hemoglobina32. Tanto los homocigóticos como los heterocigóticos parecen estar protegidos contra la malaria severa33,34 pero el efecto protector parece ser superior en los s. En estudio desarrollado en Burkina Faso35, se estimó que el efecto protector de la variante HbAC era de 30%, mientras que el de la variante HbCC era de 90%.
El efecto protector de la HbC no parece deberse a una reducción en la densidad parasitaria, y sí a una alteración en la topografía y en las propiedades de la superficie de los eritrocitos infectados involucrados en la patogenicidad36,37,38. Los eritrocitos infectados HbAC y HbCC presentan menor adhesión al endotelio que expresa CD36 y la molécula de adhesión intercelular-1 (ICAM-1) que los eritrocitos infectados HbAA. La formación de rosetas con los eritrocitos no infectados también es menor. Se observaron igualmente, alteraciones en la expresión del importante factor de virulencia del parásito PfEMP-1 en los eritrocitos infectados HbAC y HbCC. La hemoglobina C puede, así, proteger contra la malaria a través de la reducción de la adhesión de los eritrocitos infectados que es mediada por la PfEMP-1, suavizando los efectos de su secuestro en los microvasos36. Una citoadhesión disminuida parece, de esta manera, constituir un mecanismo de protección común a ambas variantes de la hemoglobina S y C.
Un estudio reciente39 abordó este tema desde otra perspectiva, cuestionando si el genotipo humano de la β-globina podría influenciar la eficiencia de la transmisión de malaria por P. falciparum. En este estudio, llevado a cabo en Burkina Faso (África Occidental), se verificó que las HbC y HbS están asociadas a un aumento de dos veces in vivo y cuatro veces ex vivo de la transmisión del parásito del huésped humano para el vector, mostrando que la variación genética humana puede influenciar en la dinámica de la transmisión de una enfermedad infecciosa. Estas variantes podrán promover la diferenciación sexual de P. falciparum o accionar respuestas inmunes alternativas que mejoran la eficiencia de la transmisión del huésped vertebrado para el vector o, al contrario, reducen la eficiencia del bloqueo de la transmisión en el huésped.
LAS TALASEMIAS
Las α- y β- talasemias son una consecuencia de deleciones y mutaciones puntuales en zonas no codificantes de los genes que codifican para las α- y β-globinas, causando una síntesis insuficiente de estas cadenas (revisto en Weatherall40).
La α-talasemia es uno de los desórdenes genéticos humanos más comunes del globo y es particularmente frecuente en los países del Mediterráneo, Sudeste Asiático, continente Africano, Oriente Medio e India41. En algunas regiones, la frecuencia de portadores llega a ser de 80% a 90% de la población, o sea, alcanza casi un estado de fijación42,43. Los individuos con mutación en un único cromosoma, con una anemia poco acentuada, son designados portadores del rasgo talasémico α y son, de manera general, asintomáticos. Los heterocigóticos compuestos y homocigóticos presentan una anemia relativamente severa caracterizada por la presencia de hemoglobina H (HbH) en la sangre periférica (enfermedad HbH). Los individuos que producen una cantidad muy reducida de globina α o no producen del todo estas cadenas, tienen una anemia muy grave que, no siendo tratada, causa la muerte en el período neonatal (condición designada de hidropesía fetal de la hemoglobina Bart). La gran mayoría de las α-talasemias resultan de la deleción de un (-α) o ambos (--) genes a pero pueden tener también origen en mutaciones puntuales. Cuando una mutación (o varias) suprime completamente la expresión de un gen, la enfermedad es llamada de α0-talasemia; cuando una o varias mutaciones reducen parcialmente la expresión de un gen, se designa de α+-talasemia (Harteveld e Higgs41 y referencias aquí contenidas).
La β-talasemia incluye tres formas principales en orden creciente de gravedad: talasemia Mayor (referida muchas veces como anemia de Cooley y anemia del Mediterráneo), talasemia intermedia y talasemia Menor (también designada de rasgo β-talasémico o β-talasemia heterocigótica). La β-talasemia tiene prevalencia en los países del Mediterráneo, Medio Oriente, Asia Central, India, Sur de China y Extremo Oriente. Las frecuencias de portadores más elevadas se encuentran en Chipre (14%), Cerdeña (10,3%) y Sudeste de Asia (Galanello y Origa44 y referencias aquí contenidas).
El hecho de las talasemias ser tan frecuentes se justifica porque los portadores están en presumible ventaja selectiva en las áreas donde la malaria por P. falciparum es o fue endémica. Varios estudios apoyan una asociación entre la reducida producción de las cadenas de hemoglobina y esta enfermedad infecciosa45,46,47.
Un estudio en Liberia, por ejemplo, determinó que había un riesgo relativo de infecciones graves de malaria, de 0,41 a 0,45, en niños heterocigóticos de β-talasemia48 y en Papúa Nueva Guinea, en un estudio de caso control comparando niños con malaria grave a niños no infectados, se concluyó que había un riesgo reducido de contraer malaria grave en los homocigóticos de α+-talasemia (riesgo de 0,40) y también en los heterocigóticos (riesgo de 0,66)49. Por otra parte, en otro estudio de caso control en Gana, los resultados indicaron que la protección contra la malaria clínica severa sucede en individuos heterocigóticos para la α+-talasemia, pero no en individuos talasémicos homocigóticos50. En otro estudio en el Sudoeste Asiático se obtuvieron resultados que van contra los resultados anteriores: los porcentajes de malaria no grave y esplenomegalia fueron mayores en los niños con α+-talasemia que en los niños normales. Este resultado fue más relevante en niños con edad muy precoz e infectados por P. vivax. En este caso, la talasemia puede estar asociada a un aumento de la susceptibilidad a P. vivax que, actuando como una vacuna natural, induce una protección contra la infección subsiguiente, potencialmente más grave, por P. falciparum26.
Para las formas menos severas de talasemias no ha surgido todavía una evidencia consistente sobre un posible efecto en la invasión o maduración del parásito. Sin embargo, algunos resultados han destacado alteraciones sutiles en el eritrocito talasémico infectado, incluyendo el aumento de la expresión de antígenos en la superficie y el aumento de la conexión de la IgG, que puede conducir a un reconocimiento inmunitario más eficiente y a una mejor remoción de los eritrocitos infectados y, de esta forma, a un mejor control de la infección en los estadios eritrocíticos51,52. Un aumento de la susceptibilidad de los eritrocitos talasémicos infectados a la fagocitosis por los monocitos fue también observada in vitro53. Ambas talasemias muestran una disminución de la formación de rosetas, un problema asociado a formas severas de malaria54.
LAS DEFICIENCIAS ENZIMÁTICAS DEL ERITROCITO
Debido a la pérdida del núcleo, mitocondrias y ribosomas, los eritrocitos maduros no tienen la capacidad de realizar ni la fosforilación oxidativa ni la síntesis proteica. Sin embargo, estas células necesitan de un metabolismo activo para el mantenimiento de la flexibilidad y la integridad de la membrana plasmática, bien como para el mantenimiento de la hemoglobina en su forma funcional para asegurar el adecuado transporte del oxígeno. Este metabolismo está asegurado por las enzimas del glóbulo rojo que participan en tres cadenas metabólicas principales: la glucólisis, la vía de las pentosas fosfato y el metabolismo nucleotídico. Si existe alguna deficiencia enzimática en alguna de estas vías, hay una limitación de la producción de ATP y/o de NADPH provocando alteraciones en la membrana y consecuente remoción de estas células55.
La relación entre el grado de la deficiencia enzimática y la extensión de la disfunción metabólica depende de varios factores: la relevancia de la enzima afectada y su grado de expresión, la estabilidad de la enzima mutante contra la degradación proteolítica y las anormalidades funcionales y la posibilidad de compensar la deficiencia por la sobreexpresión de la isoenzima correspondiente o por el uso de una vía metabólica alternativa55.
Ya han sido identificadas enzimopatías en las varias cadenas metabólicas y las frecuencias varían con la localización geográfica. De todas, la deficiencia de glucosa-6-fosfato deshidrogenasa parece ser la más común, con más de 400 millones de casos registrados. En el África subsahariana existen principalmente tres variantes que presentan frecuencias polimórficas, que fueron asociadas a una protección contra la malaria.
Más recientemente, han sido publicados algunos trabajos que asocian la deficiencia de piruvato quinasa con una protección contra la malaria. Se obtuvieron evidencias de esta asociación en modelo de ratones, estudios in vitro con cultivos de P. falciparum y estudios poblacionales con muestras de ADN humano en los que fueron identificadas marcas de selección en la región del genoma que envuelve el gen codificante de piruvato quinasa. No han sido todavía identificadas variantes de este gen con frecuencias polimórficas en las regiones endémicas de malaria, pero esta investigación está en curso.
LA DEFICIENCIA DE GLUCOSA-6-FOSFATO DESIDROGENASA
La glucosa-6-fosfato deshidrogenasa (G6PD) cataliza la primera reacción de la vía de las pentosas fosfato en la cual ocurre la oxidación de la glucosa-6-fosfato la 6-fosfoglucono-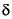 -lactona, simultáneamente a la producción de NADPH. El NADPH es un equivalente reductor, necesario a varias reacciones biosintéticas y extremadamente importante en la protección de las células contra el estrés oxidativo56,57. El gen que codifica para la G6PD se halla en la región telomérica del brazo largo (Xq28) del cromosoma X y está constituido por 13 exones que codifican un total de 515 aminoácidos y 12 intrones. El locus de la G6PD es uno de los loci más polimórficos descritos, con mm más de 300 variantes genéticas conocidas, que originan cerca de 140 variantes moleculares diferentes58. Estas variantes resultan, sobre todo, de mutaciones puntuales, no habiendo referencias de la existencia de grandes deleciones en el gen de la G6PD o de mutaciones nonsense o frameshift59.
-lactona, simultáneamente a la producción de NADPH. El NADPH es un equivalente reductor, necesario a varias reacciones biosintéticas y extremadamente importante en la protección de las células contra el estrés oxidativo56,57. El gen que codifica para la G6PD se halla en la región telomérica del brazo largo (Xq28) del cromosoma X y está constituido por 13 exones que codifican un total de 515 aminoácidos y 12 intrones. El locus de la G6PD es uno de los loci más polimórficos descritos, con mm más de 300 variantes genéticas conocidas, que originan cerca de 140 variantes moleculares diferentes58. Estas variantes resultan, sobre todo, de mutaciones puntuales, no habiendo referencias de la existencia de grandes deleciones en el gen de la G6PD o de mutaciones nonsense o frameshift59.
En el África subsahariana existen tres variantes que presentan frecuencias polimórficas: G6PD B, G6PD A y G6PD A-. La variante G6PD B es la variante normal; la variante G6PD A resulta de una mutación en la posición 376 del exón 5, en donde se hace la sustitución de una adenina (A) por una guanina (G), que resulta en un cambio de aminoácido (Asn>Asp). La variante G6PD A- se caracteriza por la ocurrencia de dos mutaciones: la descrita para la variante G6PD A y otra en el nucleótido 202 del exón 4, habiendo sustitución de una guanina (G) por una adenina (A), originando un cambio en el aminoácido codificado (Val>Met)56,57,60. La variante normal, G6PD B, presenta una actividad enzimática normal y es la más común: su frecuencia en la población varía entre 60% y 80%. La variante G6PD A, que presenta una actividad entre 80% y 100% (cuando comparada con la variante G6PD B) se halla en una frecuencia entre 15% y 40%. La G6PD A- presenta una actividad de 10 a 20% en individuos homo o hemicigóticos y su frecuencia varía entre 0 y 25%56,61.
La sintomatología presentada por los enfermos depende del grado de deficiencia, aunque gran parte de los individuos deficientes es asintomática, desarrollando síntomas de la enfermedad apenas como respuesta al estrés oxidativo. Los síntomas clínicos más comunes son: ictericia neonatal y anemia hemolítica aguda provocada por la ingestión de fármacos (como por ejemplo el antimalárico primaquina) o ciertos alimentos (como habas, de ahí que esta enfermedad sea conocida también como favismo o fabismo)56,57.
En todo el mundo cerca de 400 millones de personas son afectadas por esta enzimopatía, lo que la vuelve una de las enzimopatías más comunes56,62. No obstante, la distribución geográfica de la deficiencia de G6PD presenta mayores prevalencias en África y en algunos países de Asia, en donde la prevalencia de malaria es o fue muy elevada. Esta co-distribución puede justificarse por la hipótesis que la deficiencia de G6PD puede conferir una protección parcial contra la malaria. Aunque el grado de protección conferida y cuales sean los individuos (mujeres heterocigóticas y/u hombres hemicigóticos) que disfrutan esa protección sean blanco de controversia, parece cierto que su elevada prevalencia se debe al hecho de que existió una selección positiva a lo largo del tiempo3,22,63. Ejemplos de esta controversia pueden ser hallados en diversos estudios. Powell et al64 y Martin et al65 no observaron evidencias de ningún tipo de protección contra la infección por P. falciparum en afroamericanos y en la población nigeriana, respectivamente. Por otro lado, Bienzle et al (1972) (in Ruwende et al66), en un estudio realizado con niños nigerianos, concluyeron que apenas las mujeres heterocigóticas, y no los hombres hemicigóticos, estaban protegidos contra la malaria. Ruwende et al66, en un estudio realizado en Gambia y en Kenia, verificaron que el alelo G6PD A- confería protección contra la malaria severa tanto en mujeres heterocigóticas como en hombres hemicigóticos. Otro resultado discrepante fue obtenido por Guindo et al67, en un estudio realizado con dos poblaciones étnicamente diferentes de Malí. Los resultados obtenidos sugieren que existe una protección contra la malaria severa en hombres hemicigóticos pero no en mujeres heterocigóticas. También Gilles et al (1967) (in Guindo et al67), en un estudio realizado en Nigeria, observaron que el alelo G6PD A- tenía un efecto protector contra el coma y convulsiones en niños con edades comprendidas entre los 6 meses y los 4 años de edad. Sin embargo, los resultados encontrados no fueron estadísticamente significativos en el caso de las niñas.
LA DEFICIENCIA DE PIRUVATO QUINASA
La piruvato quinasa está involucrada en el último paso de la vía glicolítica de las células, conduciendo a la producción de piruvato y ATP. Por su vez, su substrato fosfoenolpiruvato (PEP) y el producto piruvato están involucrados en varias cadenas energéticas y biosintéticas, por lo que la regulación de la actividad de esta enzima es crucial para el metabolismo global de las células68. En los mamíferos existen cuatro isoenzimas de la piruvato quinasa (PK-M1, PK-M2, PK-L y PK-R) codificadas por dos genes distintos, pkM y pkLR, y expresados en tejidos diferentes. El gen pkLR está localizado en el cromosoma 1 (1q21) y codifica las proteínas PK-L y PK-R. La PK-L se expresa en el hígado, intestino delgado y corteza renal; la PK-R se expresa exclusivamente en los eritrocitos. La región codificante del gen pkLR está dividida en 12 exones, diez de los cuales son compartidos por las dos isoformas, mientras que los exones 1 y 2 son específicos para las enzimas eritrocitaria y hepática, respectivamente. Hasta el momento, ya han sido descritas más de 180 mutaciones y ocho polimorfismos en el gen pkLR69.
A pesar de las alteraciones en el gen pkLR pueden resultar en modificaciones, sea en la enzima del eritrocito, sea en la enzima del hígado, los síntomas clínicos se deben a las alteraciones en los glóbulos rojos, una vez que la deficiencia hepática es compensada, generalmente, por la síntesis continua en los hepatocitos. La deficiencia de PK, tal como la deficiencia de G6PD, se constituye en una enfermedad hemolítica no esferocítica hereditaria. Las manifestaciones clínicas incluyen anemias hemolíticas crónicas de varios grados, variando de poco severas a formas que deben ser compensadas por transfusiones de sangre en el período neonatal69.
La asociación de la deficiencia de PK con la malaria ha sido descrita muy recientemente. El primer estudio sobre esta asociación se realizó en 2003 por Min-Oo y colaboradores70, que observaron que dos estirpes congénitas recombinantes de ratones eran resistentes a la infección por Plasmodium chabaudi, e identificaron la mutación 269T>A en el exón 3 del gen pkLR (90lle>Asn) como inductora de la resistencia, con significativa reducción en la parasitemia y en la mortalidad pos infección. Posteriormente, ya en 2008, dos trabajos fueron publicados comparando el crecimiento de cultivos in vitro de Plasmodium en eritrocitos humanos normales y con la deficiencia71,72, observándose una disminución en la infección y en la replicación del parásito en estos últimos. Ya este año, se conocieron otros dos estudios, esta vez usando muestras de ADN humano. Alves y colaboradores73 realizaron un estudio poblacional usando muestras de ADN humano de Cabo Verde, observando un desequilibrio de conexión en una extensión mayor de la región envolvente del gen pkLR en los individuos control no infectados con malaria. Este resultado fue explorado después por el mismo equipo de investigación74, que efectuó un segundo estudio poblacional, en el cual, por medio del genotipado combinado de varios polimorfismos localizados en la proximidades del gen pkLR en muestras de ADN humano de diferentes grupos clínicos de malaria de Angola y Mozambique e individuos PK-deficientes y normales de Portugal, obtuvo los siguientes resultados: una mayor diferenciación entre los países africanos y Portugal cuando se usaron marcadores de la región pkLR comparativamente con marcadores neutros; mayor conservación de la región envolvente del gen pkLR en el grupo clínico de malaria no complicada (desequilibrio de conexión en una región más extensa); asociación positiva de un haplotipo con este grupo clínico. Estas observaciones sostienen la hipótesis de que la malaria está ejerciendo presión en esta región específica del genoma y fortalecen los resultados anteriores, obtenidos con cultivos in vitro y con modelo de ratones. En este contexto, una cuestión se coloca ahora: ¿existirá un alelo mutante de pkLR prevalente en las regiones endémicas de malaria, tal como sucede en la deficiencia de G6PD o HbS? La respuesta a esta cuestión es relevante para esta área de estudio y la identificación de uno (o varios) alelo(s) en estas condiciones vendría a confirmar que el gen pkLR está bajo selección.
ANTÍGENO DUFFY/RECEPTOR DE QUIMIOCINAS (DARC)
Plasmodium vivax, la segunda especie de Plasmodium más prevalente en el mundo, infectando entre 80 y 90 millones de personas por año75, aunque esté ampliamente distribuido en los países tropicales, es prácticamente inexistente en África Central y Occidental. Esta ausencia se ha explicado por la falta del antígeno Duffy en la mayor parte de la población76. El antígeno Duffy, también llamado de antígeno Duffy receptor para quimiocinas (DARC), una vez que este antígeno se conecta a una serie de quimiocinas proinflamatorias77,78, ha sido descrito como el receptor eritrocítico para el P. vivax sin el cual no es posible haber invasión del eritrocito por parte de este parásito.
DARC es una proteína de la membrana/tisular multimérica que está organizada en siete dominios transmembranas. El gen DARC es bastante polimórfico, presentando múltiples alelos, entre ellos los alelos codominantes FY*A y FY*B, que codifican para dos alelos principales - Fya y Fyb. Por medio de la combinación de los dos alelos principales es posible obtener cuatro genotipos diferentes: Fy(a+b+), Fy(a+b-), Fy(a-b+) y Fy(a-b-)77,78,79. Los tres primeros corresponden al fenotipo Duffy positivo, que es más frecuente en Asia y en poblaciones caucasianas, y el último corresponde al fenotipo Duffy negativo, más común en la población africana, siendo descrito como resistente a la infección por P. vivax.
El fenotipo Fy(a-b-) resulta de una única mutación puntual, la -33T>C, en la región promotora del alelo FY*B, situada en la región de "GATA box", que va a impedir la conexión del factor de transcripción h-GATA177,80. Varios estudios a lo largo de los años, han demostrado que la ausencia del antígeno Duffy impide la invasión del eritrocito por P. vivax, como es el caso del estudio realizado por Miller et al81, que demostró que la resistencia al P. vivax estaba directamente asociada al fenotipo Duffy negativo. Otro estudio realizado por Barnwell et al82 demostró in vitro que merozoitos de P. vivax son incapaces de invadir eritrocitos que no expresen el antígeno Duffy. Más recientemente, Culleton et al83, en un estudio en que analizaron 2.588 muestras de sangre de nueve países de África, encontraron apenas una muestra infectada con P. vivax en un individuo Duffy positivo. Este estudio confirmó que esta especie de Plasmodium es prácticamente inexistente en África.
Otro aspecto importante de destacar en lo que dice a la protección conferida por el antígeno Duffy y que fue descubierto por Kasehagen et al84, en un estudio realizado en Papua, Nueva Guinea, es que, además de que los individuos homocigótigos Duffy negativos están protegidos contra la invasión del P. vivax, también los individuos heterocigóticos portadores de un nuevo alelo Duffy negativo [Fy(A+A-)], que presenta una disminución de 50% en la expresión de Fya, están significativamente más protegidos contra infecciones por P. vivax que individuos homocigóticos [Fy(A+A+)], y, cuando infectados, presentan parasitemias significativamente más bajas cuando comparadas con las de individuos normales.
Estudios recientes han demostrado que hay individuos Duffy negativos infectados con P. vivax en Brasil, pero también en África. Un estudio realizado en Kenia, con niños que estaban siendo analizados para un estudio caso-control de malaria grave provocada por P. falciparum, confirmó la existencia de niños infectadas con P. vivax aún siendo Duffy negativas . Este no fue el único relato y resultados semejantes se encontraron en la Región Amazónica en Brasil, en donde también fueron detectados individuos Duffy-negativos infectados con P. vivax86,87, y también en otras localidades de África Occidental. Un estudio caso-control que está siendo desarrollado en Guinea Ecuatorial, encontró nueve individuos Duffy-negativos, conteniendo la mutación -33T>C, en la región promotora del alelo FY*B, situada en la región de la "GATA box", infectados con diferentes estirpes de P. vivax - P. vivax clásico y P. vivax VK247 (Mendesi et al, observaciones no publicadas). Estos datos recientes sugieren que P. vivax puede estar evolucionando, usando receptores alternativos para conectarse e invadir el eritrocito.
GRUPOS SANGUÍNEOS ABO
El sistema ABO es uno de los sistemas sanguíneos sobre el cual se tiene un mayor conocimiento y también uno de los más relevantes en la práctica clínica, en lo que dice respecto a la compatibilidad entre grupos sanguíneos. Este es un sistema autosómico, por lo que cada persona posee dos copias del gen que codifica para el grupo sanguíneo ABO. Los grupos sanguíneos A y B son dominantes en relación al grupo O, mientras que los grupos A y B son codominantes88.
El gen codifica una glicotransferasa, que, en el caso del grupo sanguíneo A, transfiere una N-acetil D-galactosamina y, en el caso del grupo sanguíneo B, una D-galactosa para la extremidad del glicano de las glicoproteínas o los glicolípidos88. El grupo sanguíneo O no presenta la glicotransferasa terminal, necesaria para la producción de los antígenos A y/o B, expresando el antígeno H88,89.
Diversos estudios sugieren que también el sistema ABO haya sufrido un proceso de selección positiva en humanos, por acción de la malaria. Aunque existan relatos controvertidos, con estudios que afirman que no hay una asociación significativa entre los dos factores90,91,92,93, varios , otros afirman exactamente lo contrario89,94,95,96,97,98. Analizando los resultados obtenidos en los diferentes estudios, se puede constatar que factores como: la no utilización de un grupo control o la utilización de un grupo control inadecuado; el hecho de que muchas veces se utilizan adultos para estos estudios, y no niños con edades hasta 5 años (grupo más informativo, una vez que es este el que normalmente presenta densidades parasitarias más elevadas y donde hay una mayor incidencia de las formas graves de la enfermedad); y el bajo número de muestras utilizadas por estudio; pueden influenciar las conclusiones finales99. De ser analizados los estudios que llevan en consideración todos los factores antes referidos, parece entonces que el grupo sanguíneo O protege realmente contra la malaria severa78,99,100.
Aunque no sea totalmente conocido el mecanismo de protección utilizado por el grupo O, varios estudios han destacado la formación de rosetas como el mecanismo más probable. La formación de rosetas se caracteriza por la conexión de eritrocitos infectados por P. falciparum a eritrocitos no infectados, formando una aglomeración de células, que se piensa contribuye a la patología de la malaria, una vez que provoca la obstrucción de los vasos, impidiendo la corriente sanguínea. Estudios realizados por Carlson et al101 y Udomsangpetch et al102 demostraron que las rosetas formadas con los grupos sanguíneos A, B y AB eran más grandes y más resistentes que las formadas por el grupo sanguíneo O. Se piensa que el grupo O forma menos rosetas por no tener antígenos A y B, ya que estos antígenos son receptores en los eritrocitos no infectados103.
La formación de rosetas contribuye directamente con la severidad de la enfermedad, una vez que provoca isquemia de los tejidos y muerte celular, y si este es reducido en individuos del grupo sanguíneo O, es de esperarse entonces que estos individuos estuvieran de cierta forma protegidos contra la malaria grave. Rowe et al89 demostraron por medio de un estudio caso-control realizado en Malí con niños con malaria grave y niños con malaria no complicada, que individuos del grupo sanguíneo O tenían un 66% menos probabilidades de desarrollar malaria grave cuando comparados con grupos sanguíneos no-O. Resultados similares fueron hallados por Fry et al104 en un estudio realizado en Kenia, Malawi y en Gambia, en el cual fueron genotipados más de 9 mil individuos, utilizando cuatro polimorfismos de base única (SNPs) en el gen de la glicotransferasa ABO. En este estudio se verificó que los grupos sanguíneos A y AB (odds ratios significativos de 1,33 y 1,59, respectivamente) están asociados a un riesgo mayor de malaria grave cuando comparados al grupo O.
Resultados interesantes se obtuvieron también en un estudio realizado con madres y bebés en Gambia, en donde se verificó que el grupo O está asociado a un aumento de la infección por malaria placentaria en mujeres primíparas, pero que presenta un riesgo reducido en mujeres multíparas97. Esta diferencia en la susceptibilidad a la malaria en mujeres embarazadas fue hallado solamente en el grupo sanguíneo O.
Si el grupo sanguíneo O realmente protege contra la malaria grave, sería de esperarse que presentase prevalencias más elevadas, por lo menos en las áreas endémicas para la enfermedad. Estudios afirman que las prevalencias de este grupo sanguíneo no son más elevadas porque, a pesar de tener un papel protector contra la malaria grave, los individuos del grupo O tienden a tener síntomas más graves de otras enfermedades, como el cólera y otras enfermedades diarreicas, muy comunes en países tropicales105,106.
CONCLUSIÓN
Hay una estrecha relación entre huésped y parásito que, si prolongada en el tiempo, tiende a afinarse, permitiendo que cause menor daño al huésped y traga mayor beneficio al parásito. La muerte precoz no resulta en descendencia y, por lo tanto, no trae beneficio desde el punto de vista evolutivo para ninguna de las partes. Los polimorfismos del eritrocito protectores contra la malaria en los humanos son un excelente ejemplo de esta coevolución huésped-parásito, pues las elevadas prevalencias en las zonas endémicas de malaria muestran que han sido seleccionados a lo largo del tiempo por permitir una mayor supervivencia del huésped humano y, consecuentemente, su transmisión a las generaciones siguientes.
Por otra parte, desde un punto de vista clínico, es extremamente importante evaluar los determinantes de susceptibilidad a la infección y enfermedad por malaria y comprender los mecanismos funcionales involucrados, para que puedan ser utilizados como nuevos objetivos para fármacos o vacunas. Las enzimas de las cadenas metabólicas, por ejemplo, parecen ser un mecanismo de defensa contra la infección y/o enfermedad, dada la elevada prevalencia de ciertas variantes de la G6PD (y posiblemente de la PK) en humanos, y en el parásito han sido referidas como un blanco prometedor para nuevos fármacos anti-Plasmodium, dado que las enzimas parasitarias parecen diferir, sea bioquímica, sea estructuralmente, de las congéneres del huésped.
Además, se sabe que el uso de determinados fármacos (como algunos antimaláricos) pueden, por ejemplo, desencadenar graves crisis de hemólisis en individuos G6PD deficientes, por lo que es de suma importancia el uso de técnicas de diagnóstico apropiadas para prevenir el agravo del estado clínico de los pacientes.
El conjunto de los aspectos antes mencionados muestra que el conocimiento acumulado en los últimos 60 años sobre este tema es extremamente importante y útil y puede abrir puertas para un tratamiento más eficaz para la malaria.
APOYO FINANCIERO
Se agradece la financiación atribuida por la Fundación para la Ciencia y Tecnología del Ministerio de Ciencia, Tecnología y Enseñanza Superior de Portugal [Becas SFRH/BD/28236/2006 (PM) y SFRH/BD/41473/2007 (CM)].
REFERENCIAS
1 World Health Organization. World Malaria Report. Geneva: WHO; 2008. http://apps.who.int/malaria/wmr2008/malaria2008.pdf.
2 Parikh S, Dorsey G, Rosenthal PJ. Host polymorphisms and the incidence of malaria in Ugandan children. Am J Trop Med Hyg. 2004 Dec;71(6):750-3. [Link]
3 Kwiatkowski DP. How malaria has affected the human genome and what human genetics can teach us about malaria. Am J Hum Genet. 2005 Aug;77(2):171-92. [Link]
4 Williams TN. Red blood cell defects and malaria. Mol Biochem Parasitol. 2006 Oct;149(2):121-7. DOI:10.1016/j.molbiopara.2006.05.007 [Link]
5 Wellems TE, Hayton K, Fairhurst RM. The impact of malaria parasitism: from corpuscles to communities. J Clin Invest. 2009 Sep;119(9):2496-505. DOI:10.1172/JCI38307 [Link]
6 Greenwood B, Marsh K, Snow R. Why do some African children develop severe malaria? Parasitol Today. 1991 Oct;7(10):277-81. DOI:10.1016/0169-4758(91)90096-7 [Link]
7 Rowe AK, Rowe SY, Snow RW, Korenromp EL, Schellenberg JR, Stein C, et al. The burden of malaria mortality among African children in the year 2000. Int J Epidemiol. 2006 Jun;35(3):691-704. DOI:10.1093/ije/dyl027 [Link]
8 Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature. 2005 Mar;434(7030):214-7. DOI:10.1038/nature03342 [Link]
9 Riley EM, Ong CS, Olerup O, Eida S, Allen SJ, Bennett S, et al. Cellular and humoral immune responses to Plasmodium falciparum gametocyte antigens in malaria-immune individuals: limited response to the 48/45-kilodalton surface antigen does not appear to be due to MHC restriction. J Immunol. 1990 Jun;144(12):4810-6. [Link]
10 Modiano D, Petrarca V, Sirima BS, Nebie I, Diallo D, Esposito F, et al. Different response to Plasmodium falciparum malaria in west African sympatric ethnic groups. Proc Natl Acad Sci U S A. 1996 Nov;93(23):13206-11. [Link]
11 Haldane JBS. Disease and evolution. Ric Sci. 1949;35 Suppl:S68-76.
12 Jepson A, Sisay-Joof F, Banya W, Hassan-King M, Frodsham A, Bennett S, et al. Genetic linkage of mild malaria to the major histocompatibility complex in Gambian children: study of affected sibling pairs. BMJ. 1997 Jul;315(7100):96-7. [Link]
13 Mackinnon MJ, Gunawardena DM, Rajakaruna J, Weerasingha S, Mendis KN, Carter R. Quantifying genetic and nongenetic contributions to malarial infection in a Sri Lankan population. Proc Natl Acad Sci U S A. 2000 Nov;97(23):12661-6. [Link]
14 Makinnon MJ, Mwangi TW, Snow RW, Marsh K, Williams TN. Heritability of malaria in Africa. PloS Med. 2005 Dec;2(12):e340. DOI:10.1371/journal.pmed.0020340 [ Link]
15 Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79(8):704-12. [Link]
16 Ohashi J, Naka I, Patarapotikul J, Hananantachai H, Brittenham G, Looareesuwan S, et al. Extended linkage disequilibrium surrounding the hemoglobin E variant due to malarial selection. Am J Hum Genet. 2004 Jan;74(6):1198-208. [Link]
17 Chebloune Y, Pagnier J, Trabuchet G, Faure C, Verdier G, Labie D, et al. Structural analysis of the 5' flanking region of the beta-globin gene in African sickle cell anemia patients: further evidence for three origins of the sickle cell mutation in Africa. Proc Natl Acad Sci U S A. 1988 Jun;85(12):4431-5. [Link]
18 Stuart MJ, Nagel RL. Sickle-cell disease. Lancet. 2004 Oct;364(9442):1343-60. DOI:10.1016/S0140-6736(04)17192-4 [Link]
19 Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997 Sep;337(11):762-9. [Link]
20 Allison AC. Protection afforded by sickle-cell trait against subtertian malareal infection. Br Med J. 1954 Feb;1(4857):290-4. [Link]
21 Weatherall DJ, Clegg JB. Genetic variability in response to infection: malaria and after. Genes Immun. 2002 Sep;3(6):331-7. DOI:10.1038/sj.gene.6363878 [Link]
22 Min-Oo G, Gros P. Erythrocyte variants and the nature of their malaria protective effect. Cell Microbiol. 2005 Jun;7(6):753-63. DOI:10.1111/j.1462-5822.2005.00524.x [Link]
23 Aidoo M, Terlouw DJ, Kolczak MS, McElroy PD, ter Kuile FO, Kariuki S, et al. Protective effects of the sickle cell gene against malaria morbidity and mortality. Lancet. 2002 Apr;359(9314):1311-2. DOI:10.1016/S0140-6736(02)08273-9 [Link]
24 Carter R, Mendis KN. Evolutionary and historical aspects of the burden of malaria. Clin Microbiol Rev. 2002 Oct;15(4):564-94. DOI:10.1128/CMR.15.4.564-594.2002 [Link]
25 Ayi K, Turrini F, Piga A, Arese P. Enhanced phagocytosis of ring-parasitized mutant erythrocytes: a common mechanism that may explain protection against falciparum malaria in sickle trait and beta-thalassemia trait. Blood. 2004 Nov;104(10):3364-71. DOI 10.1182/blood-2003-11-3820. [Link]
26 Williams TN, Maitland K, Bennett S, Ganczakowski M, Peto TE, Newbold CI, et al. High incidence of malaria in alpha-thalassaemic children. Nature. 1996 Oct;383(6600):522-5. DOI:10.1038/383522a0 [Link]
27 Pattanapanyasat K, Yongvanitchit K, Tongtawe P, Tachavanich K, Wanachiwanawin W, Fucharoen S, et al. Impairment of Plasmodium falciparum growth in thalassemic red blood cells: further evidence by using biotin labeling and flow cytometry. Blood. 1999 May;93(9):3116-9. [Link]
28 Cappadoro M, Giribaldi G, O'Brien E, Terrain F, Mannu F, Ulliers D, et al. Early phagocytosis of glucose-6-phosphate dehydrogenase (G6PD)- deficient erythrocytes parasitized by Plasmodium falciparum may explain malaria protection in G6PD deficiency. Blood. 1998 Oct;92(7):2527-34. [Link]
29 Williams TN, Mwangi TW, Wambua S, Alexander ND, Kortok M, Snow RW, et al. Sickle cell trait and the risk of Plasmodium falciparum malaria and other childhood diseases. J Infect Dis. 2005 Jul;192(1):178-86. DOI:10.1086/430744 [ Link]
30 Cholera R, Brittain NJ, Gillrie MR, Lopera-Mesa TM, Diakite SA, Arie T, et al. Impaired cytoadherence of Plasmodium falciparum-infected erythrocytes containing sickle hemoglobin. Proc Natl Acad Sci U S A. 2008 Jan;105(3):991-6. DOI:10.1073/pnas.0711401105 [Link]
31 Hassan DA, Arez AP, Mohamed HS, Elhussein AM, Ibrahim ME, Abdulhadi NH. The reduced sequestration of Plasmodium-falciparum-infected erythrocytes among malaria cases with sickle-cell trait: in-vivo evidence from Sudan. Ann Trop Med Parasitol. 2008 Dec;102(8):743-8. [Link]
32 Diallo DA, Doumbo OK, Dicko A, Guindo A, Coulibaly D, Kayentao K. A comparison of anemia in hemoglobin C and normal hemoglobin A children with Plasmodium falciparum malaria. Acta Trop. 2004 May;90(3):295-9. DOI:10.1016/J.ACTATROPICA.2004.02.005. [Link]
33 Agarwal A, Guindo A, Cissoko Y, Taylor JG, Coulibaly D, Kone A, et al. Hemoglobin C associated with protection from severe malaria in the Dogon of Mali, a West African population with a low prevalence of hemoglobin S. Blood. 2000 Oct;96(7):2358-63. [Link]
34 Mockenhaupt FP, Ehrhardt S, Cramer JP, Otchwemah RN, Anemana SD, Goltz K, et al. Hemoglobin C and resistance to severe malaria in Ghanaian children. J Infect Dis. 2004 Sep;190(5):1006-9. DOI:10.1086/422847. [Link]
35 Modiano D, Luoni G, Sirima BS, Simpore J, Verra F, Konate A, et al. Haemoglobin C protects against clinical Plasmodium falciparum malaria. Nature. 2001 Nov;414(6861):305-8. DOI:10.1038/35104556 [Link]
36 Fairhurst RM, Baruch DI, Brittain NJ, Ostera GR, Wallach JS, Hoang HL, et al. Abnormal display of PfEMP-1 on erythrocytes carrying haemoglobin C may protect against malaria. Nature. 2005 Jun;435(7045):1117-21. DOI:10.1038/nature03631 [Link]
37 Tokumasu F, Fairhurst RM, Ostera GR, Brittain NJ, Hwang J, Wellems TE, et al. Band 3 modifications in Plasmodium falciparum-infected AA and CC erythrocytes assayed by autocorrelation analysis using quantum dots. J Cell Sci. 2005 Mar;118(Pt 5):1091-8. DOI:10.1242/jcs.01662 [Link]
38 Arie T, Fairhurst RM, Brittain NJ, Wellems TE, Dvorak JA. Hemoglobin C modulates the surface topography of Plasmodium falciparum-infected erythrocytes. J Struct Biol. 2005 May;150(2):163-9. DOI:10.1016/j.jsb.2005.02.008 [Link]
39 Gouagna LC, Bancone G, Yao F, Yameogo B, Dabire KR, Costantini C, et al. Genetic variation in human HBB is associated with Plasmodium falciparum transmission. Nat Genet. 2010 Apr;42(4):328-31. DOI:10.1038/ng.554 [Link]
40 Weatherall DJ. Phenotype-genotype relationships in monogenic disease: lessons from the thalassaemias. Nat Rev Genet. 2001 Apr;2(4):245-55. DOI:10.1038/35066048 [Link]
41 Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis. 2010 May;5:13. DOI:1186/1750-1172-5-13 [Link]
42 Fodde R, Losekoot M, van den Broek MH, Oldenburg M, Rashida N, Schreuder A, et al. Prevalence and molecular heterogeneity of alfa+thalassemia in two tribal populations from Andhra Pradesh, India. Hum Genet. 1988 Oct;80(2):157-60. [Link]
43 Modiano G, Morpurgo G, Terrenato L, Novelletto A, Di Rienzo A, Colombo B, et al. Protection against malaria morbidity: near-fixation of the alpha-thalassemia gene in a Nepalese population. Am J Hum Genet. 1991 Feb;48(2):390-7. [Link]
44 Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010 May;5:11. [Link]
45 Flint J, Hill AV, Bowden DK, Oppenheimer SJ, Sill PR, Serjeantson SW, et al. High frequencies of alpha-thalassaemia are the result of natural selection by malaria. Nature. 1986 Jan;321(6072):744-50. [Link]
46 Hill AV. The population genetics of alpha-thalassemia and the malaria hypothesis. Cold Spring Harb Symp Quant Biol. 1986;51(Pt 1):489-98.
47 Hill AV, Bowden DK, O'Shaughnessy DF, Weatherall DJ, Clegg JB. Beta thalassemia in Melanesia: association with malaria and characterization of a common variant (IVS-1 nt 5 G----C). Blood. 1988 Jul;72(1):9-14. [Link]
48 Willcox M, Björkman A, Brohult J. Falciparum malaria and beta-thalassaemia trait in northern Liberia. Ann Trop Med Parasitol. 1983 Aug;77(4):335-47. [Link]
49 Allen SJ, O'Donnell A, Alexander ND, Alpers MP, Peto TE, Clegg JB, et al. α+-Thalassemia protects children against disease caused by other infections as well as malaria. Proc Natl Acad Sci U S A. 1997 Dec; 94(26):14736-41. [Link]
50 Mockenhaupt FP, Ehrhardt S, Gellert S, Otchwemah RN, Dietz E, Anemana SD, et al. α+-thalassemia protects African children from severe malaria. Blood. 2004 Oct;104(7):2003-6. DOI 10.1182/blood-2003-11-4090. [Link]
51 Luzzi GA, Merry AH, Newbold CI, Marsh K, Pasvol G, Weatherall DJ. Surface antigen expression on Plasmodium falciparum-infected erythrocytes is modified in a- and ß-thalassemia. J Exp Med. 1991 Apr;173(4):785-91. [Link]
52 Williams TN, Weatherall DJ, Newbold CI. The membrane characteristics of Plasmodium falciparum-infected and -uninfected heterozygous a0thalassaemic erythrocytes. Br J Haematol. 2002 Aug;118(2):663-70. [Link]
53 Yuthavong Y, Bunyaratvej A, Kamchonwongpaisan S. Increased susceptibility of malaria-infected variant erythrocytes to the mononuclear phagocyte system. Blood Cells. 1990;16(2-3):591-7. [Link]
54 Carlson J, Nash GB, Gabutti V, al-Yaman F, Wahlgren M. Natural protection against severe Plasmodium falciparum malaria due to impaired rosette formation. Blood. 1994 Dec;84(11):3909-14. [Link]
55 Jacobasch G, Rapoport SM. Hemolytic anemias due to erythrocyte enzyme deficiencies. Mol Aspects Med. 1996 Apr;17(2):143-70. [Link]
56 Ruwende C, Hill A. Glucose-6-phosphate dehydrogenase deficiency and malaria. J Mol Med. 1998 Jul;76(8):581-8. [Link]
57 Mehta A, Mason PJ, Vulliamy TJ. Glucoses-phosphate dehydrogenase deficiency. Baillieres Best Pract Res Clin Haematol. 2000 Mar;13(1):21-38. [Link]
58 Beutler E, Vulliamy TJ. Hematologically important mutations: glucose-6-phosphate dehydrogenase. Blood Cells Mol Dis. 2002 Mar-Apr;28(2):93-103. [Link]
59 Vulliamy T, Mason P, Luzzatto L. The molecular basis of glucose-6-phosphate dehydrogenase deficiency. Trends Genet. 1992 Apr;8(4):138-43. DOI:10.1016/0168-9525(92)90372-B [Link]
60 Mombo LE, Ntoumi F, Bisseye C, Ossari S, Lu CY, Nagel RL, et al. Human genetic polymorphisms and asymptomatic Plasmodium falciparum malaria in Gabonese schoolchildren. Am J Trop Med Hyg. 2003 Feb;68(2):186-90. [Link]
61 May J, Meyer CG, Grossterlinden L, Ademowo OG, Mockenhaupt FP, Olumese PE, et al. Red cell glucose-6-phosphate dehydrogenase status and pyruvate kinase activity in a Nigerian population. Trop Med Int Health. 2000 Feb;5(2):119-23. DOI:10.1046/j.1365-3156.2000.00529.x [Link]
62 Kaneko A, Taleo G, Kalkoa M, Yaviong J, Reeve PA, Ganczakowski M, et al. Malaria epidemiology, glucose 6-phosphate dehydrogenase deficiency and human settlement in the Vanuatu Archipelago. Acta Trop. 1998 Jul;70(3):285-302. DOI:10.1016/S0001-706X(98)00035-7 [Link]
63 Fortin A, Stevenson MM, Gros P. Susceptibility to malaria as a complex trait: big pressure from a tiny creature. Hum Mol Genet. 2002 Oct;11(20):2469-78. DOI:1093/hmg/11.20.2469 [Link]
64 Powell RD, Brewer GJ. Glucose-6-phosphate dehydrogenase deficiency and falciparum malaria. Am J Trop Med Hyg. 1965 May;14:358-62.
65 Martin SK, Miller LH, Alling D, Okoye VC, Esan GJ, Osunkoya BO, et al. Severe malaria and glucose-6-phosphate-dehydrogenase deficiency: a reappraisal of the malaria/G-6-PD. hypothesis. Lancet. 1979 Mar;1(8115):524-6. [ Links ]
66 Ruwende C, Khoo SC, Snow RW, Yates SN, Kwiatkowski D, Gupta S, et al. Natural selection of hemi- and heterozygotes for G6PD deficiency in Africa by resistance to severe malaria. Nature. 1995 Jul;376(6537):246-9. DOI:10.1038/376246A0 [ Links ]
67 Guindo A, Fairhurst RM, Doumbo OK, Wellems TE, Diallo DA. X-linked G6PD deficiency protects hemizygous males but not heterozygous females against severe malaria. PLoS Med. 2007 Mar;4(3):e66. [ Links ]
68 Valentini G, Chiarelli LR, Fortin R, Dolzan M, Galizzi A, Abraham DJ, et al. Structure and function of human erythrocyte pyruvate kinase. Molecular basis of nonspherocytic hemolytic anemia. J Biol Chem. 2002 Jun;277(26):23807-14. DOI:10.1074/jbc.M202107200 [ Links ]
69 Zanella A, Fermo E, Bianchi P, Chiarelli LR, Valentini G. Pyruvate kinase deficiency: the genotype-phenotype association. Blood Rev. 2007 Jul;21(4):217-31. [ Links ]
70 Min-Oo G, Fortin A, Tam MF, Nantel A, Stevenson MM, Gros P. Pyruvate kinase deficiency in mice protects against malaria. Nat Genet. 2003 Dec;35(4): 357-62. DOI: 10.1038/ng1260 [Link]
71 Ayi K, Min-Oo G, Serghides L, Crockett M, Kirby-Allen M, Quirt I, et al. Pyruvate kinase deficiency and malaria. N Engl J Med. 2008 Apr;358(17):1805-10. [Link]
72 Durand PM, Coetzer TL. Pyruvate kinase deficiency protects against malaria in humans. Haematologica. 2008 Jun;93(6):939-40. [Link]
73 Alves J, Machado P, Silva J, Gonçalves N, Ribeiro L, Faustino P, et al. Analysis of malaria associated genetic traits in Cabo Verde, a melting pot of European and sub Saharan settlers. Blood Cells Mol Dis. 2010 Jan;44(1):62-8. DOI:10.1016/j.bcmd.2009.09.008 [Link]
74 Machado P, Pereira R, Rocha AM, Manco L, Fernandes N, Miranda J, et al. Malaria: looking for selection signatures in the human PKLR gene region. Br J Haematol. 2010 Jun;149(5):775-84. DOI:10.1111/j.1365-2141.2010.08165.x [Link]
75 Souza-Neiras WC, Melo LM, Machado RL. The genetic diversity of Plasmodium vivax: a review. Mem Inst Oswaldo Cruz. 2007 Jun;102(3):245-54. [Link]
76 Langhi DM Jr, Bordin JO. Duffy blood group and malaria. Hematology. 2006 Oct;11(5):389-98. [Link]
77 Tournamille C, Colin Y, Cartron JP, Le Van Kim C. Disruption of a GATA motif in the Duffy gene promoter abolishes erythroid gene expression in Duffy-negative individuals. Nat Genet. 1995 Jun;10(2):224-8. DOI:10.1038/ng0695-224 [Link]
78 Castilho L, Rios M, Pellegrinno J Jr, Saad STO, Costa FF, Reid ME. A novel FY allele in Brazilians. Vox Sang. 2004 Oct;87(3):190-5. DOI:10.1111/j.1423-0410.2004.00554.x [Link]
79 Rowe JA, Opi DH, Williams TN. Blood groups and malaria: fresh insights into pathogenesis and identification of targets for intervention. Curr Opin Hematol. 2009 Nov;16(6):480-7. [Link]
80 Carvalho TL, Ribolla PEM, Curi RA, Mota LS. Characterization and transcriptional analysis of the promoter region of the Duffy blood group, chemokine receptor (DARC) in cattle. Vet Immunol Immunopathol. 2009 Dec;132(2-4):153-9. DOI:10.1016/j.vetimm.2009.05.016 [Link]
81 Allison AC. Genetic control of resistance to human malaria. Curr Opin Immunol. 2009 Oct;21(5):499-505. [Link]
82 Miller LH, Mason SJ, Dvorak JA, McGinniss MH, Rothman IK. Erythrocyte receptors for (Plasmodium knowlesi) malaria: Duffy blood group determinants. Science. 1975 Aug;189(4202):561-3. [Link]
83 Barnwell JW, Nichols ME, Rubinstein P. In vitro evaluation of the role of the Duffy blood group in erythrocyte invasion by Plasmodium vivax. J Exp Med. 1989 May;169(5):1795-802. [Link]
84 Culleton RL, Mita T, Ndounga M, Unger H, Cravo PV, Paganotti GM, et al. Failure to detect Plasmodium vivax in West and Central Africa by PCR species typing. Malar J. 2008 Sep;7:174. DOI:10.1186/1475-2875-7-174 [Link]
85 Kasehagen LJ, Mueller I, Kiniboro B, Bockarie MJ, Reeder JC, Kazura JW, et al. Reduced Plasmodium vivax erythrocyte infection in PNG Duffy-negative heterozygotes. PLoS One. 2007 Mar;2(3):e336. [Link]
86 Ryan JR, Stoute JA, Amon J, Dunton RF, Mtalib R, Koros J, et al. Evidence for transmission of Plasmodium vivax among a duffy antigen negative population in Western Kenya. Am J Trop Med Hyg. 2006 Oct;75(4):575-81. [Link]
87 Cavasini CE, Rossit AR , Couto AA, Couto VS, Gollino Y, Moretti LJ, et al. Duffy blood group gene polymorphisms among malaria vivax patients in four areas of the Brazilian Amazon region. Malar J. 2007 Dec;6:167. DOI:10.1186/1475-2875-6-167 [Link]
88 Cavasini CE, Mattos LC, Couto AA, Bonini-Domingos CR, Valencia SH, Neiras WC, et al. Plasmodium vivax infection among Duffy antigen-negative individuals from the Brazilian Amazon region: an exception? Trans R Soc Trop Med Hyg. 2007 Oct;101(10):1042-4. DOI:10.1016/j.trstmh.2007.04.011 [Link]
89 Anstee DJ. The relationship between blood groups and disease. Blood. 2010 Jun;115(23):4635-43. DOI 10.1182/blood-2010-01-261859. [Link]
90 Rowe JA, Handel IG, Thera MA, Deans AM, Lyke KE, Kone A, et al. Blood group O protects against severe Plasmodium falciparum malaria through the mechanism of reduced rosetting. Proc Natl Acad Sci U S A. 2007 Oct;104(44):17471-6. DOI:10.1073/PNAS.0705390104 [Link]
91 Pant CS, Gupta DK, Sharma RC, Gautam AS, Bhatt RM. Frequency of ABO blood groups, sickle-cell haemoglobin, G-6-PD deficiency and their relation with malaria in scheduled castes and scheduled tribes of Kheda District, Gujarat. Indian J Malariol. 1992 Dec;29(4):235-9. [Link]
92 Beiguelman B, Alves FP, Moura MM, Engracia V, Nunes ACS, Heckmann MIO, et al. The association of genetic markers and malaria infection in the Brazilian western Amazonian region. Mem Inst Oswaldo Cruz. 2003 Jun;98(4):455-60. DOI:10.1590/S0074-02762003000400004 [Link]
93 Migot-Nabias F, Pelleau S, Watier L, Guitard J, Toly C, Ngom MI, et al. Red blood cell polymorphisms in relation to Plasmodium falciparum asymptomatic parasite densities and morbidity in Senegal. Microbes Infect. 2006 Aug;8(9-10):2352-8. DOI:10.1016/J.MICINF.2006.03.021 [Link]
94 Uneke CJ, Ogbu O, Nwojiji V. Potential risk of induced malaria by blood transfusion in South-eastern Nigeria. Mcgill J Med. 2006 Jan;9(1):8-13. [Link]
95 Fischer PR, Boone P. Short report: severe malaria associated with blood group. Am J Trop Med Hyg. 1998 Jan;58(1):122-3. [Link]
96 Lell B, May J, Schmidt-Ott RJ, Lehman LG, Luckner D, Greve B, et al. The role of red blood cell polymorphisms in resistance and susceptibility to malaria. Clin Infect Dis. 1999 Apr;28(4):794-9. [Link]
97 Pathirana SL, Alles HK, Bandara S, Phone-Kyaw M, Perera MK, Wickremasinghe AR, et al. ABO-blood-group types and protection against severe, Plasmodium falciparum malaria. Ann Trop Med Parasitol. 2005 Mar;99(2):119-24. DOI:10.1179/136485905X19946 [Link]
98 Loscertales MP, Brabin BJ. ABO phenotypes and malaria related outcomes in mothers and babies in The Gambia: a role for histo-blood groups in placental malaria? Malar J. 2006 Aug;5:72. DOI:10.1186/1475-2875-5-72 [Link]
99 Uneke CJ. Plasmodium falciparum malaria and ABO blood group: is there any relationship? Parasitol Res. 2007 Mar;100(4):759-65. DOI:10.107/s00436-006-0342-5 [Link]
100 Cserti CM, Dzik WH. The ABO blood group system and Plasmodium falciparum malaria. Blood. 2007 Oct;110(7):2250-8. DOI:10.1182/blood-2007-03-077602. [Link]
101 Loscertales MP, Owens S, O'Donnell J, Bunn J, Bosch-Capblanch X, Brabin BJ. ABO blood group phenotypes and Plasmodium falciparum malaria: unlocking a pivotal mechanism. Adv Parasitol. 2007;65:1-50. DOI:10.1016/S0065-308X(07)65001-5 [Link]
102 Carlson J, Wahlgren M. Plasmodium falciparum erythrocyte rosetting is mediated by promiscuous lectin-like interactions. J Exp Med. 1992 Nov;176(5):1311-7. [Link]
103 Udomsangpetch R, Todd J, Carlson J, Greenwood BM. The effects of hemoglobin genotype and ABO blood group on the formation of rosettes by Plasmodium falciparum-infected red blood cells. Am J Trop Med Hyg. 1993 Feb;48(2):149-53. [Link]
104 Barragan A, Kremsner PG, Wahlgren M, Carlson J. Blood group A antigen is a coreceptor in Plasmodium falciparum rosetting. Infect Immun. 2000 May;68(5):2971-5. [Link]
105 Fry AE, Griffiths MJ, Auburn S, Diakite M, Forton JT, Green A, et al. Common variation in the ABO glycosyltransferase is associated with susceptibility to severe Plasmodium falciparum malaria. Hum Mol Genet. 2008 Feb;17(4):567-76. DOI:10.1093/hmg/ddm331 [Link]
106 van Loon FP, Clemens JD, Sack DA, Rao MR, Ahmed F, Chowdhury S, et al. ABO blood groups and the risk of diarrhea due to enterotoxigenic Escherichia coli. J Infect Dis. 1991 Jun;163(6):1243-6. [Link]
107 Swerdlow DL, Mintz ED, Rodriguez M, Tejada E, Ocampo C, Espejo L, et al. Severe life-threatening cholera associated with blood group O in Peru: implications for the Latin American epidemic. J Infect Dis. 1994 Aug;170(2):468-72.
 Correspondência / Correspondence / Correspondencia:
Correspondência / Correspondence / Correspondencia:
Patrícia Machado
Unidade de Ensino e Investigação de Malária, Instituto de Higiene e
Medicina Tropical
Rua da Junqueira n° 100, 1349-008 Lisboa-Portugal
Tel.:+351 21 365 26 00 (ext. 308)
E-mail: pmachado@ihmt.unl.pt
Recebido em / Received / Recibido en: 27/10/2010
Aceito em / Accepted / Aceito en: 16/12/2010
*Contribuíram igualmente para o presente trabalho.