








Serviços Personalizados
Journal

Artigo
 texto em
texto em  Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Pan-Amazônica de Saúde
versão impressa ISSN 2176-6215versão On-line ISSN 2176-6223
Rev Pan-Amaz Saude v.1 n.4 Ananindeua dez. 2010
http://dx.doi.org/10.5123/S2176-62232010000400013
ARTIGO DE REVISÃO | REVIEW ARTICLE | ARTÍCULO DE REVISIÓN
A contribuição dos polimorfismos humanos do eritrócito na proteção contra a malária
The contribution of human erythrocyte polymorphisms in the protection against malaria
La contribución de los polimorfismos humanos del eritrocito en la protección contra la malaria
Patrícia Machado*; Cristina Mendes*; Virgílio Estólio do Rosário; Ana Paula Arez
Unidade de Ensino e Investigação de Malária, Centro de Malária e outras Doenças Tropicais, Laboratório Associado, Instituto de Higiene e Medicina Tropical, Lisboa, Portugal
Endereço para correspondência
Correspondence
Dirección para correspondencia
RESUMO
A compreensão do complexo ciclo de vida da malária tem aumentado muito nos últimos anos mas, apesar de décadas de pesquisa e luta contra a doença, esta continua a ser um dos principais problemas de saúde pública, especialmente nas áreas mais pobres do planeta. Devido à sua elevada prevalência em certas regiões do globo, desde há cerca de 10 mil anos, a malária tem exercido uma pressão seletiva muito forte no genoma humano. A componente genética de suscetibilidade ao parasita é complexa, com uma variedade de polimorfismos a influenciar a patogénese e resposta do hospedeiro, e um dos desafios na luta contra esta doença é avaliar estes determinantes de suscetibilidade e decifrar os mecanismos envolvidos para utilizá-los como novos alvos para fármacos ou vacinas. Entre os polimorfismos genéticos humanos descritos como protetores contra a malária, os mais comuns e melhor caracterizados envolvem proteínas estruturais específicas (tais como as hemoglobinas S e C, as talassémias, o antigénio Duffy e o grupo sanguíneo O) e enzimas eritrocitárias (como a deficiência de glucose-6-fosfato desidrogenase, e, mais recentemente descrita, a deficiência de piruvato cinase). Esta pequena revisão aborda estas variantes genéticas e discute alguns dos resultados controversos obtidos, assim como os mecanismos que podem justificar esta proteção.
Palavras-chave: Malária; Polimorfismo Genético; Anemia Hemolítica Congênita; Hemoglobinas Anormais; Sistema do Grupo Sanguíneo Duffy; Sistema do Grupo Sanguíneo ABO.
ABSTRACT
The understanding of the complex life cycle of malaria has greatly improved in the last few years, however, despite decades of research and struggle against the disease, it continues to be a major public health problem, especially in the poorest areas of the world. Due to its long-term high prevalence in certain regions of the globe, malaria has exerted strong selective pressure on the human genome. The genetic component of malaria susceptibility is complex, with a variety of polymorphisms influencing both pathogenesis and host response. Evaluating these determinants of susceptibility and deciphering the mechanisms involved may lead to the discovery of new vaccines or targets for pharmacological agents. The most common and best characterized human genetic polymorphisms that confer protection against malaria involve specific structural erythrocyte proteins (such as haemoglobin S and C, thalassemias, the Duffy antigen, and blood group O) and enzymes (such as glucose-6-phosphate dehydrogenase deficiency and, more recently described, pyruvate kinase deficiency). This short review describes these genetic variants, reviews some of the controversial results that have been obtained, and discusses mechanisms that might explain the protection they provide.
Keywords: Malaria; Polymorphism, Genetic; Anemia, Hemolytic, Congenital; Hemoglobins, Abnormal; Duffy Blood-Group System; ABO Blood-Group System.
RESUMEN
La comprensión del complejo ciclo de vida de la malaria ha aumentado mucho en los últimos años pero, a pesar de décadas de investigación y lucha contra esa enfermedad, esta continúa a ser uno de los principales problemas de salud pública, especialmente en las áreas más pobres del planeta. Debido a su elevada prevalencia en ciertas regiones del globo, desde hace cerca de 10 mil años, la malaria ha ejercido una presión selectiva muy fuerte sobre el genoma humano. El componente genético de susceptibilidad al parásito es complejo, con una variedad de polimorfismos influyendo en la patogénesis y la respuesta del hospedero, y uno de los desafíos en la lucha contra esta enfermedad es evaluar estos determinantes de susceptibilidad y descifrar los mecanismos involucrados para utilizarlos como nuevas metas para fármacos o vacunas. Entre los polimorfismos genéticos humanos descritos como protectores contra la malaria, lo más comunes y mejor caracterizados involucran a proteínas estructurales específicas (tales como las hemoglobinas S y C, las talasemias, el antígeno Duffy y el grupo sanguíneo O) y enzimas eritrocitarias (como la deficiencia de glucosa-6-fosfato deshidrogenasa, y, más recientemente descrita, la deficiencia de piruvato quinasa). Esta pequeña revisión aborda estas variantes genéticas y discute algunos de los resultados controvertidos obtenidos, así como los mecanismos que pueden justificar esta protección.
Palabras clave: Malaria; Polimorfismo Genético; Anemia Hemolítica Congénita; Hemoglobinas Anormales; Sistema del Grupo Sanguíneo Duffy; Sistema del Grupo Sanguíneo ABO.
INTRODUÇÃO
Segundo a Organização Mundial da Saúde, a malária continua a ser uma das principais causas de morbidade e mortalidade nos países tropicais, estimando-se que, só no ano de 2008, tenha causado cerca de 243 milhões de casos clínicos que resultaram em aproximadamente 863 mil mortes1. A malária é uma doença infecciosa, causada por parasitas protozoários do género Plasmodium e transmitida aos humanos por picadas de mosquitos infectados do género Anopheles. As cinco espécies parasitárias que infectam os humanos são: Plasmodium falciparum, Plasmodium ovale, Plasmodium malariae, Plasmodium vivax e Plasmodium knowlesi. As espécies P. vivax e P. falciparum são as mais comuns, sendo esta última a responsável pelas formas graves da doença - malária cerebral ou anemia grave.
Sendo uma doença de muito elevada prevalência há milhares de anos, a malária tem exercido uma pressão seletiva no genoma humano2,3, especialmente nos eritrócitos, que desempenham um papel fundamental, como células hospedeiras, no ciclo de vida do parasita. Assim, os genes que afetam a estrutura e/ou funcionalidade dos eritrócitos são os que apresentam maior número de variantes genéticas descritas como associadas com a proteção contra a malária ou os seus sintomas4, como é o caso de algumas hemoglobinopatias, as talassémias, o antigénio Duffy, o sistema ABO, a deficiência de glucose-6-fosfato desidrogenase (G6PD) e, mais recentemente estudada, a deficiência de piruvato cinase (PK).
Nos últimos anos, novas linhas de investigação da malária têm surgido, como aquelas que se debruçam sobre a estreita relação entre o hospedeiro e o parasita, que explica, por um lado, a elevada abundância de P. falciparum em África, e por outro, a razão da maioria dos indivíduos parasitados não desenvolverem complicações de malária severa, enquanto outros sucumbem à doença5.
Nesta pequena revisão são abordados alguns dos polimorfismos do eritrócito humano, dos mais tradicionais aos mais recentemente identificados, descritos em trabalhos anteriores como protetores contra a malária, e são discutidos os resultados controversos que têm sido obtidos ao longo do tempo, assim como alguns dos possíveis mecanismos subjacentes a esta proteção. É dada especial ênfase às espécies de parasitas P. falciparum e P. vivax por serem as espécies mais comuns que infectam os humanos, sendo P. falciparum a mais mortal, e é dada especial atenção a regiões da África subsaariana, por serem as mais afetadas.
A SUSCETIBILIDADE À MALÁRIA É UMA CARACTERÍSTICA HEREDITÁRIA
A maioria das picadas de mosquitos infectados não produz doença (infecção com sintomatologia clássica de febre, dores de cabeça, entre outros). As crianças expostas a tais picadas ou não são infectadas (cerca de metade), ou têm infecção sem sintomas (cerca de 25%), ou têm infecção com febre e outros sintomas sem gravidade (aproximadamente 25%). Só em casos raros as crianças desenvolvem manifestações de malária severa como a anemia grave, o coma e a malária cerebral. Greenwood e colaboradores6 estimaram que em áreas de elevada transmissão, em 400 picadas potencialmente infecciosas, apenas 200 resultam em infecção e somente duas originam malária grave e uma, a morte.
Os fatores que determinam o desenvolvimento da infecção e da doença são, pois, importantes e devem ser clarificados. Podemos falar da combinação de diversos aspectos interligados: a taxa de inoculação do mosquito, a dose de esporozoítos, a imunidade adquirida de infecções anteriores, a virulência do parasita, os polimorfismos genéticos do hospedeiro humano, o estado de nutrição do indivíduo infectado, as condições ambientais e o acesso a um tratamento eficiente. A prevalência da infecção de P. falciparum é, apesar de tudo isto, suficientemente alta para matar 1 milhão de pessoas em África por ano, onde a taxa de mortalidade em crianças com menos de 5 anos de idade chega a ser entre 1 e 2%1,7,8.
Os fatores genéticos do hospedeiro humano dão uma contribuição significativa para a diversidade observada na malária. Dentro de uma mesma população, existe um elevado grau de variação entre indivíduos relativamente aos fenótipos de suscetibilidade à malária, incluindo a carga parasitária, a incidência da doença e a severidade6 e a magnitude e tipo de resposta imune aos antigénios da malária9,10.
Quando a base genética de algumas desordens do eritrócito foi inicialmente investigada, os cientistas se depararam com o estranho paradoxo da presença de elevadas frequências de mutações deletérias em algumas populações. A talassémia, por exemplo, que está na base de uma anemia microcítica, é muito frequente em várias regiões do Mediterrâneo, Médio Oriente, África e Sudoeste da Ásia. Haldane11, em 1949, propôs que um alelo mutado atinge e mantém uma elevada frequência, não por meio de uma excepcionalmente alta taxa de mutação, mas sim porque é uma consequência de uma vantagem seletiva contra a malária causada por P. falciparum, cuja distribuição coincide com a da talassémia.
Para avaliar o impacto da determinação genética da suscetibilidade à malária clínica, foi feito um estudo longitudinal com crianças gémeas na Gâmbia12 onde se observou que existe uma maior probabilidade de ambos os gémeos monozigóticos desenvolverem febre provocada pela malária do que gémeos dizigóticos, o que indica que há fatores genéticos envolvidos no desenvolvimento da doença. Num outro estudo desenvolvido no Sri-Lanka13, estimou-se que o fator hereditariedade é responsável por cerca de 15% da incidência de infecção sintomática e assintomática por P. falciparum e aproximadamente 10% da intensidade dos sintomas clínicos. Num estudo no Quênia14, foram monitoradas em duas coortes de crianças a incidência de malária clínica não complicada e as admissões hospitalares devido à malária. Em ambos os casos, foi estimado que 25% da variação total era explicada pela soma dos efeitos dos genes do hospedeiro e, destes, a hemoglobina S, o fator genético de resistência à malária mais conhecido, explicava apenas 2% da variação total, sugerindo a existência de muitos genes protetores desconhecidos, cada um resultando em pequenos efeitos nas populações.
AS DESORDENS DA HEMOGLOBINA
A hemoglobina normal de um adulto é composta por duas cadeias de α-globina e duas de β-globina. As desordens da hemoglobina podem ser divididas em dois grupos: um em que há uma diminuição na produção das formas estruturais normais das αou β-globinas, dando origem às αou β-talassémias, respectivamente; e uma outra em que há a produção de formas estruturais mutantes (hemoglobinopatias), como no caso das hemoglobinas S (HbS), C (HbC) ou E (HbE). Apesar de terem sido identificadas centenas de variantes estruturais da hemoglobina, apenas as três referidas anteriormente apresentam frequências polimórficas. As duas primeiras são muito frequentes especialmente na África Ocidental15 e a hemoglobina E é comum no Sudeste Asiático16.
As cadeias da β-globina são codificadas por um único gene que se localiza no cromossoma 11; as cadeias αsão codificadas por dois genes intimamente ligados no cromossoma 16. Assim, num indivíduo normal diploide, existem dois loci que codificam para a cadeia β e quatro que codificam para a cadeia α.
A DREPANOCITOSE OU ANEMIA FALCIFORME
A drepanocitose ou anemia falciforme é uma hemoglobinopatia de caráter genético, causada por uma mutação na posição 6 do gene da β-globina (HbB), havendo a substituição do ácido glutâmico por uma valina (β6Glu>Val)17,18. A forma anormal produzida, denominada hemoglobina S, faz com que os glóbulos vermelhos se tornem rígidos e com forma de foice, impedindo que estes atravessem os vasos sanguíneos e cheguem aos órgãos19. Os efeitos clínicos provocados por esta doença são bastante variáveis, dependendo de serem os indivíduos portadores do traço drepanocítico (HbAS, que apresentam o alelo normal A e o alelo mutado S) ou se são indivíduos doentes (HbSS, indivíduos homozigóticos para o alelo mutado S). Enquanto nos indivíduos portadores os sintomas da doença, quando existentes, são muito ligeiros, os indivíduos doentes apresentam sintomas graves como anemias severas, infecções graves e lesões em órgãos vitais, provocando uma diminuição na esperança média de vida4. A variante S da hemoglobina encontra-se amplamente distribuída por todo o mundo, apresentando frequências muito elevadas na África subsaariana (especialmente na região ocidental), no Médio Oriente e em algumas zonas da Índia15.
A drepanocitose foi das primeiras hemoglobinopatias a serem associadas a uma proteção contra a malária. Anthony Allison20, em 1954, constatou que os eritrócitos dos indivíduos com traço drepanocítico eram mais dificilmente parasitados por P. falciparum do que os normais, concluindo que os indivíduos heterozigóticos teriam uma vantagem seletiva nas regiões hiperendémicas de malária. Várias evidências sugerem a existência de um equilíbrio entre a eliminação do alelo mutado (alelo S) pela morte precoce dos homozigóticos e a preservação do mesmo nos heterozigóticos devido a esta vantagem seletiva. Os indivíduos portadores do alelo S parecem estar favorecidos relativamente aos indivíduos não portadores, havendo assim a seleção deste alelo, permitindo que este seja hoje encontrado numa percentagem de 5 a 40% na população mundial15,21,22.
Vários estudos têm sido realizados ao longo dos últimos 60 anos para compreender qual o nível de proteção conferido pelo estado heterozigótico. Aidoo et al23, Carter e Mendis24, Ayi et al25 e Williams et al26 demonstraram que o genótipo HbAS está significativamente associado à proteção contra a malária severa provocada pelo parasita P. falciparum. Aidoo et al23 demonstraram que havia uma proteção significativa de 60% na mortalidade em crianças entre os 2 e os 16 meses de idade. Em crianças com idades inferiores a 2 meses essa redução não foi observada. Williams et al26 demonstraram que havia uma proteção de 90% em indivíduos heterozigóticos (HbAS) em casos de malária severa e uma proteção de 50% nos casos de malária assintomática.
Existe controvérsia sobre os mecanismos subjacentes a esta proteção, mas alguns estudos associam-na com a dificuldade que os parasitas têm em invadir e crescer dentro dos eritrócitos com os genes mutados27. No entanto, outros estudos, como o de Cappadoro et al28, dizem não ter encontrado quaisquer diferenças no crescimento dos parasitas em eritrócitos normais e em eritrócitos mutados. Outra explicação foi dada por Luzzato et al (1970) e Roth et al (1978) (in Williams et al29). Estes verificaram que os eritrócitos HbAS parasitados têm tendência a ficar com uma forma irregular mais rapidamente que os não parasitados, levando à morte intracelular do parasita. Um estudo mais recente30 mostrou que a adesão de eritrócitos parasitados AS às células endoteliais microvasculares e aos monócitos é significativamente menor do que a dos eritrócitos parasitados AA. Esta redução está correlacionada com alterações no principal ligando de citoadesão e fator de virulência do parasita (P. falciparum erythrocyte membraneprotein-1, PfEMP-1).
Num outro estudo31, observou-se que a percentagem de trofozoítos no sangue periférico em crianças assintomáticas com traço drepanocítico (HbAS) era de 75%, enquanto nas crianças assintomáticas normais (HbAA) era de 37,5%, sugerindo que há um sequestro reduzido dos eritrócitos parasitados nas crianças HbAS. Nos indivíduos HbAS podem pois ocorrer alterações na expressão dos ligandos dos eritrócitos infectados (como o PfEMP-1), reduzindo a citoadesão e originando uma infecção menos grave. Estes resultados vão de encontro aos obtidos no estudo anterior30.
A HEMOGLOBINA C (HBC)
A hemoglobina C (HbC), tal como a hemoglobina S, é provocada por uma mutação na posição 6 do gene da β-globina havendo, neste caso, a substituição do ácido glutâmico por uma lisina (β6Glu>Lys). A hemoglocina C é principalmente encontrada na África Ocidental, embora seja menos frequente que a hemoglobina S, e resulta num fenótipo menos severo que a drepanocitose: os homozigóticos têm geralmente uma anemia hemolítica ligeira e os heterozigóticos não apresentam uma redução significativa nos níveis de hemoglobina32. Quer os homozigóticos quer os heterozigóticos parecem estar protegidos contra a malária severa33,34 mas o efeito protetor parece ser superior nos homozigóticos. Em um estudo desenvolvido em Burkina Faso35, estimou-se que o efeito protetor da variante HbAC era de 30%, enquanto o da variante HbCC era de 90%.
O efeito protetor da HbC não parece dever-se a uma redução na densidade parasitária, mas sim a uma alteração na topografia e nas propriedades da superfície dos eritrócitos infectados envolvidas na patogenicidade36,37,38. Os eritrócitos infectados HbAC e HbCC apresentam uma menor adesão ao endotélio que expressa CD36 e a molécula de adesão intercelular-1 (ICAM-1) do que os eritrócitos infectados HbAA. A formação de rosetas com os eritrócitos não infectados também é menor. Observaram-se igualmente alterações na expressão do importante fator de virulência do parasita PfEMP-1 nos eritrócitos infectados HbAC e HbCC. A hemoglobina C pode, assim, proteger contra a malária por meio da redução da adesão dos eritrócitos infectados que é mediada pela PfEMP-1, suavizando os efeitos do seu sequestro nos microvasos36. Uma diminuída citoadesão parece, assim, constituir um mecanismo de proteção comum a ambas as variantes da hemoglobina S e C.
Um estudo recente39 abordou este tema de outra perspectiva, questionando se o genótipo humano da β-globina poderia influenciar a eficiência da transmissão de malária por P. falciparum. Neste estudo, levado a cabo em Burkina Faso (África Ocidental), verificou-se que a HbC e HbS estão associadas a um aumento de duas vezes in vivo e quatro vezes ex vivo da transmissão do parasita do hospedeiro humano para o vetor, mostrando que a variação genética humana pode influenciar a dinâmica da transmissão de uma doença infecciosa. Estas variantes poderão promover a diferenciação sexual de P. falciparum ou acionar respostas imunes alternativas que melhoram a eficiência da transmissão do hospedeiro vertebrado para o vetor ou, pelo contrário, reduzem a eficiência no bloqueio da transmissão no hospedeiro.
AS TALASSÉMIAS
As α- e β- talassémias são uma consequência de deleções e mutações pontuais em zonas não codificantes dos genes que codificam para as α- e β- globinas, causando uma síntese insuficiente destas cadeias (revisto em Weatherall40).
A α-talassémia é uma das desordens genéticas humanas mais comuns do globo e é particularmente frequente nos países do Mediterrâneo, Sudeste Asiático, continente Africano, Médio Oriente e Índia41. Em algumas regiões, a frequência de portadores chega a ser de 80% a 90% da população, ou seja, atinge quase a fixação42,43. Os indivíduos com mutação num único cromossoma, com uma anemia pouco acentuada, são designados portadores do traço talassémico α e são, de uma maneira geral, assintomáticos. Os heterozigóticos compostos e homozigóticos apresentam uma anemia relativamente severa caracterizada pela presença de hemoglobina H (HbH) no sangue periférico (doença HbH). Os indivíduos que produzem uma quantidade muito reduzida de globina α ou não produzem de todo estas cadeias, têm uma anemia muito grave que, não sendo tratada, causa a morte no período neonatal (condição designada de hidropsia fetal da hemoglobina Bart). A grande maioria das α-talassémias resultam da deleção de um (-α) ou ambos (--) os genes αmas podem também ter origem em mutações pontuais. Quando uma mutação (ou várias) suprime completamente a expressão de um gene, a doença é chamada de α0-talassémia; quando uma ou várias mutações reduzem parcialmente a expressão de um gene, designa-se de α+-talassémia (Harteveld e Higgs41 e referências aqui contidas).
A β-talassémia inclui três formas principais por ordem crescente de gravidade: talassémia Major (referida muitas vezes como anemia de Cooley e anemia do Mediterrâneo), talassémia intermédia e talassémia Minor (também designada de traço β-talassémico ou β-talassémia em heterozigotia). A β-talassémia é prevalente nos países do Mediterrâneo, Médio Oriente, Ásia Central, Índia, Sul da China e Extremo Oriente. As mais elevadas frequências de portadores são encontradas no Chipre (14%), Sardenha (10,3%) e Sudeste da Ásia (Galanello e Origa44 e referências aqui contidas).
O fato das talassémias serem tão frequentes justifica-se pelos portadores estarem presumivelmente em vantagem seletiva nas áreas onde a malária por P. falciparum é ou foi endémica. Vários estudos suportam uma associação entre a reduzida produção das cadeias de hemoglobina e esta doença infecciosa45,46,47.
Um estudo na Libéria, por exemplo, determinou que havia um risco relativo de infecções graves de malária, de 0,41 a 0,45, em crianças heterozigóticas de β-talassémia48 e na Papua Nova Guiné, num estudo caso-controlo comparando crianças com malária grave com crianças não infectadas, concluiu-se que havia um risco reduzido de contrair malária grave nos homozigóticos de α+-talassémia (risco de 0,40) e também nos heterozigóticos (risco de 0,66)49. Por outro lado, noutro estudo caso-controlo no Gana, os resultados indicaram que a proteção contra a malária clínica severa acontece em indivíduos heterozigóticos para a α+-talassémia mas não em indivíduos talassémicos homozigóticos50. Noutro estudo no Sudoeste Asiático foram obtidos resultados que vão contra os resultados anteriores: as percentagens de malária não grave e esplenomegalia foram maiores nas crianças com α+-talassémia do que nas crianças normais. Este resultado foi mais relevante em crianças com idade muito precoce e infectadas por P. vivax. Neste caso, a talassémia poderá estar associada a um aumento da suscetibilidade a P. vivax que, atuando como uma vacina natural, induz uma proteção contra a infecção subsequente potencialmente mais grave por P. falciparum26.
Para as formas menos severas de talassémias não surgiu ainda uma evidência consistente sobre um possível efeito na invasão ou maturação do parasita. No entanto, alguns resultados têm apontado para alterações sutis no eritrócito talassémico infectado, incluindo o aumento da expressão de antigénios na superfície e aumento da ligação da IgG, que pode levar a um reconhecimento imunitário mais eficiente e uma melhor remoção dos eritrócitos infectados e, assim, um melhor controlo da infecção nos estadios eritrocíticos51,52. Um aumento da suscetibilidade dos eritrócitos talassémicos infectados à fagocitose pelos monócitos foi também observado in vitro53. Ambas as talassémias mostram uma diminuição da formação de rosetas, um problema associado com formas severas de malária54.
AS DEFICIÊNCIAS ENZIMÁTICAS DO ERITRÓCITO
Devido à perda do núcleo, mitocôndrias e ribossomas, os eritrócitos maduros não têm a capacidade de realizar nem a fosforilação oxidativa nem a síntese proteica. No entanto, estas células necessitam de um metabolismo ativo para a manutenção da flexibilidade e integridade da membrana plasmática, assim como para a manutenção da hemoglobina na sua forma funcional para assegurar o transporte adequado do oxigénio. Este metabolismo é assegurado pelas enzimas do glóbulo vermelho que participam em três cadeias metabólicas principais: a glicólise, a via das pentoses-fosfato e o metabolismo nucleotídico. Se existir alguma deficiência enzimática em alguma destas vias, há uma limitação na produção de ATP e/ou de NADPH provocando alterações na membrana e consequente remoção destas células55.
A relação entre o grau da deficiência enzimática e a extensão da disfunção metabólica depende de vários fatores: a relevância da enzima afetada e o seu grau de expressão, a estabilidade da enzima mutante contra a degradação proteolítica e as anormalidades funcionais e a possibilidade de compensar a deficiência pela sobre-expressão da isoenzima correspondente ou pelo uso de uma via metabólica alternativa55.
Já foram identificadas enzimopatias nas várias cadeias metabólicas e as frequências variam com a localização geográfica. De todas elas, a deficiência de glucose-6-fosfato desidrogenase parece ser a mais comum, com mais de 400 milhões de casos registados. Na África subsaariana existem principalmente três variantes que apresentam frequências polimórficas, que foram associadas com uma proteção contra a malária.
Mais recentemente, também têm sido publicados alguns trabalhos que associam a deficiência de piruvato cinase com uma proteção contra a malária. Foram obtidas evidências desta associação em modelo de ratinho, estudos in vitro com culturas de P. falciparum e estudos populacionais com amostras de DNA humano onde foram identificadas marcas de seleção na região do genoma que envolve o gene codificante da piruvato cinase. Não foram ainda identificadas variantes deste gene com frequências polimórficas nas regiões endémicas de malária, mas esta pesquisa está em curso.
A DEFICIÊNCIA DE GLUCOSE-6-FOSFATO DESIDROGENASE
A glucose-6-fosfato desidrogenase (G6PD) catalisa a primeira reação da via das pentoses-fosfato na qual se dá a oxidação da glucose-6-fosfato a 6-fosfoglucono-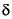 -lactona, havendo simultaneamente a produção de NADPH. O NADPH é um equivalente redutor, necessário a várias reações biossintéticas e extremamente importante na proteção das células contra o stress oxidativo56,57. O gene que codifica para a G6PD encontra-se na região telomérica do braço longo (Xq28) do cromossoma X e é constituído por 13 exões que codificam um total de 515 aminoácidos e 12 intrões. O locus da G6PD é um dos loci mais polimórficos descritos, com mais de 300 variantes genéticas conhecidas, que originam cerca de 140 variantes moleculares diferentes58. Estas variantes resultam, sobretudo, de mutações pontuais, não havendo referências da existência de grandes deleções no gene da G6PD ou de mutações nonsense ou frameshiff59.
-lactona, havendo simultaneamente a produção de NADPH. O NADPH é um equivalente redutor, necessário a várias reações biossintéticas e extremamente importante na proteção das células contra o stress oxidativo56,57. O gene que codifica para a G6PD encontra-se na região telomérica do braço longo (Xq28) do cromossoma X e é constituído por 13 exões que codificam um total de 515 aminoácidos e 12 intrões. O locus da G6PD é um dos loci mais polimórficos descritos, com mais de 300 variantes genéticas conhecidas, que originam cerca de 140 variantes moleculares diferentes58. Estas variantes resultam, sobretudo, de mutações pontuais, não havendo referências da existência de grandes deleções no gene da G6PD ou de mutações nonsense ou frameshiff59.
Na África subsaariana existem três variantes que apresentam frequências polimórficas: G6PD B, G6PD A e G6PD A-. A variante G6PD B é a variante normal; a variante G6PD A resulta de uma mutação na posição 376 do exão 5, onde há a substituição de uma adenina (A) por uma guanina (G), que resulta numa mudança de aminoácido (Asn>Asp). A variante G6PD A- caracteriza-se pela ocorrência de duas mutações: a descrita para a variante G6PD A e uma outra no nucleótido 202 do exão 4, ocorrendo a substituição de uma guanina (G) por uma adenina (A), originando uma mudança no aminoácido codificado (Val>Met)56,57,60. A variante normal, G6PD B, apresenta uma atividade enzimática normal e é a mais comum: a sua frequência na população varia entre 60% e 80%. A variante G6PD A, que apresenta uma atividade entre 80% e 100% (quando comparada com a variante G6PD B) encontra-se numa frequência entre 15% e 40%. A G6PD A- apresenta uma atividade de 10 a 20% em indivíduos homo ou hemizigóticos e a sua frequência varia entre 0 e 25%56,61.
A sintomatologia apresentada pelos doentes depende do grau de deficiência, mas grande parte dos indivíduos deficientes é assintomática, desenvolvendo sintomas da doença apenas como resposta ao stress oxidativo. Os sintomas clínicos mais comuns são: icterícia neonatal e anemia hemolítica aguda provocada pela ingestão de fármacos (como por exemplo o antimalárico primaquina) ou certos alimentos (como favas, daí esta doença ser também designada por favismo)56,57.
Em todo o mundo cerca de 400 milhões de pessoas são afetadas por esta enzimopatia, o que a torna uma das enzimopatias mais comuns56,62. Contudo, a distribuição geográfica da deficiência de G6PD apresenta as maiores prevalências na África e alguns países da Ásia, onde a prevalência de malária é ou foi muito elevada. Esta codistribuição pode ser justificada pela hipótese da deficiência de G6PD conferir uma proteção parcial contra a malária. Embora o grau de proteção conferida e quais os indivíduos (mulheres heterozigóticas e/ou homens hemizigóticos) que usufruem dessa proteção sejam alvo de controvérsia, parece certo que a sua elevada prevalência se deve ao fato de ter havido uma seleção positiva ao longo do tempo3,22,63. Exemplos desta controvérsia podem ser encontrados em diversos estudos. Powell et al64 e Martin et al65 não observaram evidências de qualquer tipo de proteção contra a infecção por P. falciparum em afroamericanos e na população nigeriana, respectivamente. Por outro lado, Bienzle et al (1972) (in Ruwende et al66), num estudo realizado com crianças nigerianas, concluíram que apenas as mulheres heterozigóticas, e não os homens hemizigóticos, estavam protegidos contra a malária. Ruwende et al66, num estudo realizado na Gâmbia e no Quênia, verificaram que o alelo G6PD A- conferia proteção contra a malária severa tanto em mulheres heterozigóticas como em homens hemizigóticos. Um outro resultado discrepante foi obtido por Guindo et al67, num estudo realizado com duas populações etnicamente diferentes do Mali. Os resultados obtidos sugerem que existe uma proteção contra a malária severa em homens hemizigóticos mas não em mulheres heterozigóticas. Também Gilles et al (1967) (in Guindo et al67), num estudo realizado na Nigéria, observaram que o alelo G6PD A- tinha um efeito protetor contra o coma e convulsões em crianças com idades compreendidas entre os 6 meses e os 4 anos de idade. Contudo, os resultados encontrados não foram estatisticamente significativos no caso das crianças do sexo feminino.
A DEFICIÊNCIA DE PIRUVATO CINASE
A piruvato cinase está envolvida no último passo da via glicolítica das células, levando à produção de piruvato e ATP. Por sua vez, o seu substrato fosfoenolpiruvato (PEP) e o produto piruvato estão envolvidos em várias cadeias energéticas e biossintéticas, pelo que a precisa regulação da atividade desta enzima é crucial para o metabolismo global das células68. Nos mamíferos existem quatro isoenzimas da piruvato cinase (PK-M1, PK-M2, PK-L e PK-R) codificadas por dois genes distintos, pkM e pkLR, e expressos em tecidos diferentes. O gene pkLR está localizado no cromossoma 1 (1q21) e codifica as proteínas PK-L e a PK-R. A PK-L é expressa no fígado, intestino delgado e cortex renal; a PK-R é exclusivamente expressa nos eritrócitos. A região codificante do gene pkLR está dividida em 12 exões, dez dos quais são partilhados pelas duas isoformas, enquanto os exões 1 e 2 são específicos para as enzimas eritrocitária e hepática, respectivamente. Até ao momento, já foram descritos mais de 180 mutações e oito polimorfismos no gene pkLR69.
Apesar das alterações no gene pkLR poderem resultar em modificações, quer na enzima do eritrócito, quer na enzima do fígado, os sintomas clínicos devem-se às alterações nos glóbulos vermelhos, uma vez que a deficiência hepática é geralmente compensada pela síntese contínua nos hepatócitos. A deficiência de PK, tal como a deficiência de G6PD, constitui uma doença hemolítica não esferocítica hereditária. As manifestações clínicas incluem anemias hemolíticas crónicas de vários graus, variando de pouco severas a formas que têm de ser compensadas por transfusões de sangue no período neonatal69.
A associação da deficiência de PK com a malária foi descrita muito recentemente. O primeiro estudo sobre esta associação foi efetuado em 2003 por Min-Oo e colaboradores70, que observaram que duas estirpes congénitas recombinantes de ratinho eram resistentes à infecção por Plasmodium chabaudi, e identificaram a mutação 269T>A no exão 3 do gene pkLR (90lle>Asn) como indutora da resistência, com significativa redução na parasitémia e na mortalidade após a infecção. Posteriormente, já em 2008, dois trabalhos foram publicados comparando o crescimento de culturas in vitro de Plasmodium em eritrócitos humanos normais e com a deficiência71,72, tendo-se observado uma diminuição na infecção e replicação do parasita nestes últimos. Já este ano, dois outros estudos foram conhecidos, desta vez usando amostras de DNA humano. Alves e colaboradores73 realizaram um estudo populacional usando amostras de DNA humano de Cabo Verde, observando um desequilíbrio de ligação numa maior extensão da região envolvente do gene pkLR nos indivíduos controlo não infectados com malária. Este resultado foi explorado depois pela mesma equipa de investigação74, que efetuou um segundo estudo populacional, no qual, por meio da genotipagem combinada de vários polimorfismos localizados na vizinhança do gene pkLR em amostras de DNA humano de diferentes grupos clínicos de malária de Angola e Moçambique e indivíduos PK-deficientes e normais de Portugal, obteve os seguintes resultados: uma maior diferenciação entre os países africanos e Portugal quando foram usados marcadores da região pkLR comparativamente com marcadores neutros; maior conservação da região envolvente do gene pkLR no grupo clínico de malária não complicada (desequilíbrio de ligação numa região mais extensa); associação positiva de um haplótipo com este grupo clínico. Estas observações sustentam a hipótese de que a malária está a exercer pressão nesta região específica do genoma e fortalecem os resultados anteriores, obtidos com culturas in vitro e com modelo de ratinho. Neste contexto, uma questão é agora colocada: existirá um alelo mutante de pkLR prevalente nas regiões endémicas de malária, tal como acontece na deficiência de G6PD ou HbS? A resposta a esta questão é relevante para esta área de estudo e a identificação de um (ou vários) alelo(s) nestas condições viria confirmar que o gene pkLR está sob seleção.
ANTIGÉNIO DUFFY/RECEPTOR DE QUEMOQUINAS (DARC)
Plasmodium vivax, a segunda espécie de Plasmodium mais prevalente no mundo, infectando entre 80 e 90 milhões de pessoas por ano75, embora esteja amplamente distribuído nos países tropicais, é praticamente inexistente em África Central e Ocidental. Esta ausência tem sido explicada pela falta do antigénio Duffy na maior parte da população76. O antigénio Duffy, também chamado de antigénio Duffy receptor para quemoquinas (DARC), uma vez que este antigénio se liga a uma série de quemoquinas pró-inflamatórias77,78, tem sido descrito como o receptor eritrocítico para o P. vivax e sem o qual não é possível haver invasão do eritrócito por parte deste parasita.
DARC é uma proteína membranar multimérica que está organizada em sete domínios transmembranares. O gene DARC é bastante polimórfico, apresentando múltiplos alelos, entre eles os alelos codominantes FY*A e FY*B, que codificam para dois alelos principais - Fya e Fyb. Por meio da combinação dos dois alelos principais é possível obter quatro genótipos diferentes: Fy(a+b+), Fy(a+b-), Fy(a-b+) e Fy(a-b-)77,78,79. Os três primeiros correspondem ao fenótipo Duffy positivo, que é mais frequente na Ásia e em populações caucasianas, e o último corresponde ao fenótipo Duffy negativo, mais comum na população africana, estando descrito como sendo resistente à infecção por P. vivax.
O fenótipo Fy(a-b-) resulta de uma única mutação pontual, a -33T>C, na região promotora do alelo FY*B, situada na região da "GATA box", que vai impedir a ligação do fator de transcrição h-GATA177,80. Vários estudos ao longo dos anos têm demonstrado que a ausência do antigénio Duffy impede a invasão do eritrócito por P. vivax, como é o caso do estudo realizado por Miller et al81, que demonstrou que a resistência ao P. vivax estava diretamente associada com o fenótipo Duffy negativo. Um outro estudo realizado por Barnwell et al82 demonstrou in vitro que merozoítos de P. vivax são incapazes de invadir eritrócitos que não expressem o antigénio Duffy. Mais recentemente, Culleton et al83, num estudo em que analisaram 2.588 amostras de sangue de nove países de África, encontraram apenas uma amostra infectada com P. vivax num indivíduo Duffy positivo. Este estudo confirmou que esta espécie de Plasmodium é praticamente inexistente em África.
Um outro aspecto que é importante realçar no que diz respeito à proteção conferida pelo antigénio Duffy e que foi descoberto por Kasehagen et al84, num estudo realizado na Papua, Nova Guiné, é que, para além dos indivíduos homozigótigos Duffy negativos estarem protegidos contra a invasão do P. vivax, também os indivíduos heterozigóticos portadores de um novo alelo Duffy negativo [Fy(A+A-)], que apresenta uma diminuição de 50% na expressão de Fya, estão significativamente mais protegidos contra infecções por P. vivax do que indivíduos homozigóticos [Fy(A+A.+)], e, quando infectados, apresentam parasitémias significativamente mais baixas quando comparadas com as de indivíduos normais.
Estudos recentes têm demonstrado que há indivíduos Duffy negativos infectados com P. vivax no Brasil, mas também em África. Um estudo realizado no Quênia, com crianças que estavam a ser analisadas para um estudo caso-controlo de malária grave provocada por P. falciparum, confirmou a existência de crianças infectadas com P. vivax mesmo sendo Duffy negativas85. Este não foi o único relato e resultados semelhantes foram encontrados na Região Amazónica no Brasil, onde também foram detectados indivíduos Duffy-negativos infectados com P. vivax86,87, e também noutras localidades de África Ocidental. Um estudo caso-controlo que está a ser desenvolvido na Guiné-Equatorial, encontrou nove indivíduos Duffy-negativos, contendo a mutação -33T>C, na região promotora do alelo FY*B, situada na região da "GATA box", infectados com diferentes estirpes de P. vivax -P. vivax clássico e P. vivax VK247 (Mendes et al, observações não publicadas). Estes dados recentes sugerem que P. vivax pode estar a evoluir, usando receptores alternativos para se ligar e invadir o eritrócito.
GRUPOS SANGUÍNEOS ABO
O sistema ABO é um dos sistemas sanguíneos sobre o qual se tem um maior conhecimento e também um dos mais relevantes na prática clínica, no que diz respeito à compatibilidade entre grupos sanguíneos. Este é um sistema autossómico, pelo que cada pessoa possui duas cópias do gene que codifica para o grupo sanguíneo ABO. Os grupos sanguíneos A e B são dominantes em relação ao grupo O, enquanto os grupos A e B são codominantes88.
O gene codifica para uma glicotransferase, que, no caso do grupo sanguíneo A, transfere uma N-acetil D-galactosamina e, no caso do grupo sanguíneo B, uma D-galactose para a extremidade do glicano das glicoproteínas ou dos glicolípidos88. O grupo sanguíneo O não apresenta a glicotransferase terminal, necessária para a produção dos antigénios A e/ou B, expressando o antigénio H88.89.
Diversos estudos sugerem que também o sistema ABO tenha sofrido um processo de seleção positiva em humanos, por ação da malária. Embora existam relatos controversos, com estudos a afirmarem que não há uma associação significativa entre os dois fatores90,91,92,93, vários outros afirmam exatamente o contrário 89,94,95,96,97,98. Analisando os resultados obtidos nos diferentes estudos, pode-se constatar que fatores como: a não utilização de um grupo controlo ou a utilização de um grupo controlo inadequado; o fato de muitas das vezes utilizarem adultos para estes estudos, e não crianças com idades até aos 5 anos (grupo mais informativo, uma vez que é este que normalmente apresenta densidades parasitárias mais elevadas e onde há uma maior incidência das formas graves da doença); e o baixo número de amostras utilizadas por estudo; podem influenciar as conclusões finais99. Se forem analisados os estudos que levam em consideração todos os fatores antes referidos, parece então que o grupo sanguíneo O protege realmente contra a malária severa78,99,100.
Embora não seja totalmente conhecido o mecanismo de proteção utilizado pelo grupo O, vários estudos têm apontado a formação de rosetas como o mecanismo mais provável. A formação de rosetas caracteriza-se pela ligação de eritrócitos infectados com P. falciparum a eritrócitos não infectados, formando uma aglomeração de células, que se pensa contribuírem para a patologia da malária, uma vez que provoca a obstrução dos vasos, impedindo a corrente sanguínea. Estudos realizados por Carlson et al101 e Udomsangpetch et al102 demonstraram que as rosetas formadas com os grupos sanguíneos A, B e AB eram maiores e mais resistentes do que as formadas pelo grupo sanguíneo O. Pensa-se que o grupo O forma menos rosetas por não possuir antigénios A e B, pois estes antigénios são receptores nos eritrócitos não infectados103.
A formação de rosetas contribui diretamente para a severidade da doença, uma vez que provoca isquemia dos tecidos e morte celular, e se este é reduzido em indivíduos do grupo sanguíneo O, é então de esperar que estes indivíduos estivessem de certa forma protegidos contra a malária grave. Rowe et al89 demonstraram por meio de um estudo caso-controlo realizado no Mali com crianças com malária grave e crianças com malária não complicada, que indivíduos do grupo sanguíneo O tinham 66% menos probabilidade de desenvolverem malária grave quando comparados com grupos sanguíneos não-O. Resultados semelhantes foram encontrados por Fry et al104 num estudo realizado no Quênia, Malawi e na Gâmbia, no qual foram genotipados mais de 9 mil indivíduos, utilizando quatro polimorfismos de base única (SNPs) no gene da glicotransferase ABO. Neste estudo verificou-se que os grupos sanguíneos A e AB (odds ratios significativos de 1,33 e 1,59, respectivamente) estão associados a um maior risco de malária grave quando comparados com o grupo O.
Resultados interessantes foram também obtidos num estudo realizado com mães e bebés na Gâmbia, onde se verificou que o grupo O está associado com um aumento da infecção por malária placentar em mulheres primíparas, mas que apresenta um risco reduzido em mulheres multíparas97. Esta diferença na suscetibilidade à malária em mulheres grávidas foi encontrada apenas no grupo sanguíneo O.
Se o grupo sanguíneo O realmente proteger contra a malária grave, seria de esperar que apresentasse prevalências mais elevadas, pelo menos nas áreas endémicas para a doença. Estudos afirmam que as prevalências deste grupo sanguíneo não são mais elevadas porque, apesar de ter um papel protetor contra a malária grave, os indivíduos do grupo O tendem a ter sintomas mais graves de outras doenças, como a cólera e outras doenças diarreicas, muito comuns em países tropicais105,106.
CONCLUSÃO
Entre hospedeiro e parasita há uma estreita relação que, sendo prolongada no tempo, tende a ser afinada, permitindo que cause menor dano ao hospedeiro e traga maior benefício para o parasita. A morte precoce não resulta em descendência e, portanto, não traz benefício do ponto de vista evolutivo para nenhuma das partes. Os polimorfismos do eritrócito protetores contra a malária nos humanos são um excelente exemplo desta coevolução hospedeiro-parasita, pois as elevadas prevalências nas zonas endémicas de malária mostram que têm sido selecionados ao longo do tempo por permitirem uma maior sobrevivência do hospedeiro humano e, consequentemente, a sua transmissão às gerações seguintes.
Por outro lado, de um ponto de vista clínico, é extremamente importante avaliar os determinantes de suscetibilidade à infecção e doença por malária e compreender os mecanismos funcionais envolvidos, para que possam vir a ser utilizados como novos alvos para fármacos ou vacinas. As enzimas das cadeias metabólicas, por exemplo, parecem ser um mecanismo de defesa contra a infecção e/ou doença, dada a elevada prevalência de certas variantes da G6PD (e possivelmente da PK) em humanos, e no parasita têm sido referidas como um alvo promissor para novos fármacos anti-Plasmodium, dado que as enzimas parasitárias parecem diferir, quer bioquimica, quer estruturalmente, das congéneres do hospedeiro.
Além disso, sabe-se que o uso de determinados fármacos (como alguns antimaláricos) podem, por exemplo, desencadear graves crises de hemólise em indivíduos G6PD deficientes, pelo que é de toda a importância o uso de técnicas de diagnóstico apropriadas para prevenir o agravamento do estado clínico dos pacientes.
O conjunto dos aspectos acima mencionados mostra que o conhecimento acumulado nos últimos 60 anos sobre este tema é extremamente importante e útil e pode abrir portas para um tratamento mais eficaz para a malária.
APOIO FINANCEIRO
Agradece-se o financiamento atribuído pela Fundação para a Ciência e Tecnologia do Ministério da Ciência, Tecnologia e Ensino Superior de Portugal [Bolsas SFRH/BD/28236/2006 (PM) e SFRH/BD/41473/2007 (CM)].
REFERENCIAS
1 World Health Organization. World Malaria Report. Geneva: WHO; 2008. http://apps.who.int/malaria/wmr2008/malaria2008.pdf.
2 Parikh S, Dorsey G, Rosenthal PJ. Host polymorphisms and the incidence of malaria in Ugandan children. Am J Trop Med Hyg. 2004 Dec;71(6):750-3. [Link]
3 Kwiatkowski DP. How malaria has affected the human genome and what human genetics can teach us about malaria. Am J Hum Genet. 2005 Aug;77(2):171-92. [Link]
4 Williams TN. Red blood cell defects and malaria. Mol Biochem Parasitol. 2006 Oct;149(2):121-7. DOI:10.1016/j.molbiopara.2006.05.007 [Link]
5 Wellems TE, Hayton K, Fairhurst RM. The impact of malaria parasitism: from corpuscles to communities. J Clin Invest. 2009 Sep;119(9):2496-505. DOI:10.1172/JCI38307 [Link]
6 Greenwood B, Marsh K, Snow R. Why do some African children develop severe malaria? Parasitol Today. 1991 Oct;7(10):277-81. DOI:10.1016/0169-4758(91)90096-7 [Link]
7 Rowe AK, Rowe SY, Snow RW, Korenromp EL, Schellenberg JR, Stein C, et al. The burden of malaria mortality among African children in the year 2000. Int J Epidemiol. 2006 Jun;35(3):691-704. DOI:10.1093/ije/dyl027 [Link]
8 Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature. 2005 Mar;434(7030):214-7. DOI:10.1038/nature03342 [Link]
9 Riley EM, Ong CS, Olerup O, Eida S, Allen SJ, Bennett S, et al. Cellular and humoral immune responses to Plasmodium falciparum gametocyte antigens in malaria-immune individuals: limited response to the 48/45-kilodalton surface antigen does not appear to be due to MHC restriction. J Immunol. 1990 Jun;144(12):4810-6. [Link]
10 Modiano D, Petrarca V, Sirima BS, Nebie I, Diallo D, Esposito F, et al. Different response to Plasmodium falciparum malaria in west African sympatric ethnic groups. Proc Natl Acad Sci U S A. 1996 Nov;93(23):13206-11. [Link]
11 Haldane JBS. Disease and evolution. Ric Sci. 1949;35 Suppl:S68-76.
12 Jepson A, Sisay-Joof F, Banya W, Hassan-King M, Frodsham A, Bennett S, et al. Genetic linkage of mild malaria to the major histocompatibility complex in Gambian children: study of affected sibling pairs. BMJ. 1997 Jul;315(7100):96-7. [Link]
13 Mackinnon MJ, Gunawardena DM, Rajakaruna J, Weerasingha S, Mendis KN, Carter R. Quantifying genetic and nongenetic contributions to malarial infection in a Sri Lankan population. Proc Natl Acad Sci U S A. 2000 Nov;97(23):12661-6. [Link]
14 Makinnon MJ, Mwangi TW, Snow RW, Marsh K, Williams TN. Heritability of malaria in Africa. PloS Med. 2005 Dec;2(12):e340. DOI:10.1371/journal.pmed.0020340 [ Link]
15 Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79(8):704-12. [Link]
16 Ohashi J, Naka I, Patarapotikul J, Hananantachai H, Brittenham G, Looareesuwan S, et al. Extended linkage disequilibrium surrounding the hemoglobin E variant due to malarial selection. Am J Hum Genet. 2004 Jan;74(6):1198-208. [Link]
17 Chebloune Y, Pagnier J, Trabuchet G, Faure C, Verdier G, Labie D, et al. Structural analysis of the 5' flanking region of the beta-globin gene in African sickle cell anemia patients: further evidence for three origins of the sickle cell mutation in Africa. Proc Natl Acad Sci U S A. 1988 Jun;85(12):4431-5. [Link]
18 Stuart MJ, Nagel RL. Sickle-cell disease. Lancet. 2004 Oct;364(9442):1343-60. DOI:10.1016/S0140-6736(04)17192-4 [Link]
19 Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997 Sep;337(11):762-9. [Link]
20 Allison AC. Protection afforded by sickle-cell trait against subtertian malareal infection. Br Med J. 1954 Feb;1(4857):290-4. [Link]
21 Weatherall DJ, Clegg JB. Genetic variability in response to infection: malaria and after. Genes Immun. 2002 Sep;3(6):331-7. DOI:10.1038/sj.gene.6363878 [Link]
22 Min-Oo G, Gros P. Erythrocyte variants and the nature of their malaria protective effect. Cell Microbiol. 2005 Jun;7(6):753-63. DOI:10.1111/j.1462-5822.2005.00524.x [Link]
23 Aidoo M, Terlouw DJ, Kolczak MS, McElroy PD, ter Kuile FO, Kariuki S, et al. Protective effects of the sickle cell gene against malaria morbidity and mortality. Lancet. 2002 Apr;359(9314):1311-2. DOI:10.1016/S0140-6736(02)08273-9 [Link]
24 Carter R, Mendis KN. Evolutionary and historical aspects of the burden of malaria. Clin Microbiol Rev. 2002 Oct;15(4):564-94. DOI:10.1128/CMR.15.4.564-594.2002 [Link]
25 Ayi K, Turrini F, Piga A, Arese P. Enhanced phagocytosis of ring-parasitized mutant erythrocytes: a common mechanism that may explain protection against falciparum malaria in sickle trait and beta-thalassemia trait. Blood. 2004 Nov;104(10):3364-71. DOI 10.1182/blood-2003-11-3820. [Link]
26 Williams TN, Maitland K, Bennett S, Ganczakowski M, Peto TE, Newbold CI, et al. High incidence of malaria in alpha-thalassaemic children. Nature. 1996 Oct;383(6600):522-5. DOI:10.1038/383522a0 [Link]
27 Pattanapanyasat K, Yongvanitchit K, Tongtawe P, Tachavanich K, Wanachiwanawin W, Fucharoen S, et al. Impairment of Plasmodium falciparum growth in thalassemic red blood cells: further evidence by using biotin labeling and flow cytometry. Blood. 1999 May;93(9):3116-9. [Link]
28 Cappadoro M, Giribaldi G, O'Brien E, Terrain F, Mannu F, Ulliers D, et al. Early phagocytosis of glucose-6-phosphate dehydrogenase (G6PD)- deficient erythrocytes parasitized by Plasmodium falciparum may explain malaria protection in G6PD deficiency. Blood. 1998 Oct;92(7):2527-34. [Link]
29 Williams TN, Mwangi TW, Wambua S, Alexander ND, Kortok M, Snow RW, et al. Sickle cell trait and the risk of Plasmodium falciparum malaria and other childhood diseases. J Infect Dis. 2005 Jul;192(1):178-86. DOI:10.1086/430744 [ Link]
30 Cholera R, Brittain NJ, Gillrie MR, Lopera-Mesa TM, Diakite SA, Arie T, et al. Impaired cytoadherence of Plasmodium falciparum-infected erythrocytes containing sickle hemoglobin. Proc Natl Acad Sci U S A. 2008 Jan;105(3):991-6. DOI:10.1073/pnas.0711401105 [Link]
31 Hassan DA, Arez AP, Mohamed HS, Elhussein AM, Ibrahim ME, Abdulhadi NH. The reduced sequestration of Plasmodium-falciparum-infected erythrocytes among malaria cases with sickle-cell trait: in-vivo evidence from Sudan. Ann Trop Med Parasitol. 2008 Dec;102(8):743-8. [Link]
32 Diallo DA, Doumbo OK, Dicko A, Guindo A, Coulibaly D, Kayentao K. A comparison of anemia in hemoglobin C and normal hemoglobin A children with Plasmodium falciparum malaria. Acta Trop. 2004 May;90(3):295-9. DOI:10.1016/J.ACTATROPICA.2004.02.005. [Link]
33 Agarwal A, Guindo A, Cissoko Y, Taylor JG, Coulibaly D, Kone A, et al. Hemoglobin C associated with protection from severe malaria in the Dogon of Mali, a West African population with a low prevalence of hemoglobin S. Blood. 2000 Oct;96(7):2358-63. [Link]
34 Mockenhaupt FP, Ehrhardt S, Cramer JP, Otchwemah RN, Anemana SD, Goltz K, et al. Hemoglobin C and resistance to severe malaria in Ghanaian children. J Infect Dis. 2004 Sep;190(5):1006-9. DOI:10.1086/422847. [Link]
35 Modiano D, Luoni G, Sirima BS, Simpore J, Verra F, Konate A, et al. Haemoglobin C protects against clinical Plasmodium falciparum malaria. Nature. 2001 Nov;414(6861):305-8. DOI:10.1038/35104556 [Link]
36 Fairhurst RM, Baruch DI, Brittain NJ, Ostera GR, Wallach JS, Hoang HL, et al. Abnormal display of PfEMP-1 on erythrocytes carrying haemoglobin C may protect against malaria. Nature. 2005 Jun;435(7045):1117-21. DOI:10.1038/nature03631 [Link]
37 Tokumasu F, Fairhurst RM, Ostera GR, Brittain NJ, Hwang J, Wellems TE, et al. Band 3 modifications in Plasmodium falciparum-infected AA and CC erythrocytes assayed by autocorrelation analysis using quantum dots. J Cell Sci. 2005 Mar;118(Pt 5):1091-8. DOI:10.1242/jcs.01662 [Link]
38 Arie T, Fairhurst RM, Brittain NJ, Wellems TE, Dvorak JA. Hemoglobin C modulates the surface topography of Plasmodium falciparum-infected erythrocytes. J Struct Biol. 2005 May;150(2):163-9. DOI:10.1016/j.jsb.2005.02.008 [Link]
39 Gouagna LC, Bancone G, Yao F, Yameogo B, Dabire KR, Costantini C, et al. Genetic variation in human HBB is associated with Plasmodium falciparum transmission. Nat Genet. 2010 Apr;42(4):328-31. DOI:10.1038/ng.554 [Link]
40 Weatherall DJ. Phenotype-genotype relationships in monogenic disease: lessons from the thalassaemias. Nat Rev Genet. 2001 Apr;2(4):245-55. DOI:10.1038/35066048 [Link]
41 Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis. 2010 May;5:13. DOI:1186/1750-1172-5-13 [Link]
42 Fodde R, Losekoot M, van den Broek MH, Oldenburg M, Rashida N, Schreuder A, et al. Prevalence and molecular heterogeneity of alfa+thalassemia in two tribal populations from Andhra Pradesh, India. Hum Genet. 1988 Oct;80(2):157-60. [Link]
43 Modiano G, Morpurgo G, Terrenato L, Novelletto A, Di Rienzo A, Colombo B, et al. Protection against malaria morbidity: near-fixation of the alpha-thalassemia gene in a Nepalese population. Am J Hum Genet. 1991 Feb;48(2):390-7. [Link]
44 Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010 May;5:11. [Link]
45 Flint J, Hill AV, Bowden DK, Oppenheimer SJ, Sill PR, Serjeantson SW, et al. High frequencies of alpha-thalassaemia are the result of natural selection by malaria. Nature. 1986 Jan;321(6072):744-50. [Link]
46 Hill AV. The population genetics of alpha-thalassemia and the malaria hypothesis. Cold Spring Harb Symp Quant Biol. 1986;51(Pt 1):489-98.
47 Hill AV, Bowden DK, O'Shaughnessy DF, Weatherall DJ, Clegg JB. Beta thalassemia in Melanesia: association with malaria and characterization of a common variant (IVS-1 nt 5 G----C). Blood. 1988 Jul;72(1):9-14. [Link]
48 Willcox M, Björkman A, Brohult J. Falciparum malaria and beta-thalassaemia trait in northern Liberia. Ann Trop Med Parasitol. 1983 Aug;77(4):335-47. [Link]
49 Allen SJ, O'Donnell A, Alexander ND, Alpers MP, Peto TE, Clegg JB, et al. α+-Thalassemia protects children against disease caused by other infections as well as malaria. Proc Natl Acad Sci U S A. 1997 Dec; 94(26):14736-41. [Link]
50 Mockenhaupt FP, Ehrhardt S, Gellert S, Otchwemah RN, Dietz E, Anemana SD, et al. α+-thalassemia protects African children from severe malaria. Blood. 2004 Oct;104(7):2003-6. DOI 10.1182/blood-2003-11-4090. [Link]
51 Luzzi GA, Merry AH, Newbold CI, Marsh K, Pasvol G, Weatherall DJ. Surface antigen expression on Plasmodium falciparum-infected erythrocytes is modified in a- and ß-thalassemia. J Exp Med. 1991 Apr;173(4):785-91. [Link]
52 Williams TN, Weatherall DJ, Newbold CI. The membrane characteristics of Plasmodium falciparum-infected and -uninfected heterozygous a0thalassaemic erythrocytes. Br J Haematol. 2002 Aug;118(2):663-70. [Link]
53 Yuthavong Y, Bunyaratvej A, Kamchonwongpaisan S. Increased susceptibility of malaria-infected variant erythrocytes to the mononuclear phagocyte system. Blood Cells. 1990;16(2-3):591-7. [Link]
54 Carlson J, Nash GB, Gabutti V, al-Yaman F, Wahlgren M. Natural protection against severe Plasmodium falciparum malaria due to impaired rosette formation. Blood. 1994 Dec;84(11):3909-14. [Link]
55 Jacobasch G, Rapoport SM. Hemolytic anemias due to erythrocyte enzyme deficiencies. Mol Aspects Med. 1996 Apr;17(2):143-70. [Link]
56 Ruwende C, Hill A. Glucose-6-phosphate dehydrogenase deficiency and malaria. J Mol Med. 1998 Jul;76(8):581-8. [Link]
57 Mehta A, Mason PJ, Vulliamy TJ. Glucoses-phosphate dehydrogenase deficiency. Baillieres Best Pract Res Clin Haematol. 2000 Mar;13(1):21-38. [Link]
58 Beutler E, Vulliamy TJ. Hematologically important mutations: glucose-6-phosphate dehydrogenase. Blood Cells Mol Dis. 2002 Mar-Apr;28(2):93-103. [Link]
59 Vulliamy T, Mason P, Luzzatto L. The molecular basis of glucose-6-phosphate dehydrogenase deficiency. Trends Genet. 1992 Apr;8(4):138-43. DOI:10.1016/0168-9525(92)90372-B [Link]
60 Mombo LE, Ntoumi F, Bisseye C, Ossari S, Lu CY, Nagel RL, et al. Human genetic polymorphisms and asymptomatic Plasmodium falciparum malaria in Gabonese schoolchildren. Am J Trop Med Hyg. 2003 Feb;68(2):186-90. [Link]
61 May J, Meyer CG, Grossterlinden L, Ademowo OG, Mockenhaupt FP, Olumese PE, et al. Red cell glucose-6-phosphate dehydrogenase status and pyruvate kinase activity in a Nigerian population. Trop Med Int Health. 2000 Feb;5(2):119-23. DOI:10.1046/j.1365-3156.2000.00529.x [Link]
62 Kaneko A, Taleo G, Kalkoa M, Yaviong J, Reeve PA, Ganczakowski M, et al. Malaria epidemiology, glucose 6-phosphate dehydrogenase deficiency and human settlement in the Vanuatu Archipelago. Acta Trop. 1998 Jul;70(3):285-302. DOI:10.1016/S0001-706X(98)00035-7 [Link]
63 Fortin A, Stevenson MM, Gros P. Susceptibility to malaria as a complex trait: big pressure from a tiny creature. Hum Mol Genet. 2002 Oct;11(20):2469-78. DOI:1093/hmg/11.20.2469 [Link]
64 Powell RD, Brewer GJ. Glucose-6-phosphate dehydrogenase deficiency and falciparum malaria. Am J Trop Med Hyg. 1965 May;14:358-62.
65 Martin SK, Miller LH, Alling D, Okoye VC, Esan GJ, Osunkoya BO, et al. Severe malaria and glucose-6-phosphate-dehydrogenase deficiency: a reappraisal of the malaria/G-6-PD. hypothesis. Lancet. 1979 Mar;1(8115):524-6. [ Links ]
66 Ruwende C, Khoo SC, Snow RW, Yates SN, Kwiatkowski D, Gupta S, et al. Natural selection of hemi- and heterozygotes for G6PD deficiency in Africa by resistance to severe malaria. Nature. 1995 Jul;376(6537):246-9. DOI:10.1038/376246A0 [ Links ]
67 Guindo A, Fairhurst RM, Doumbo OK, Wellems TE, Diallo DA. X-linked G6PD deficiency protects hemizygous males but not heterozygous females against severe malaria. PLoS Med. 2007 Mar;4(3):e66. [ Links ]
68 Valentini G, Chiarelli LR, Fortin R, Dolzan M, Galizzi A, Abraham DJ, et al. Structure and function of human erythrocyte pyruvate kinase. Molecular basis of nonspherocytic hemolytic anemia. J Biol Chem. 2002 Jun;277(26):23807-14. DOI:10.1074/jbc.M202107200 [ Links ]
69 Zanella A, Fermo E, Bianchi P, Chiarelli LR, Valentini G. Pyruvate kinase deficiency: the genotype-phenotype association. Blood Rev. 2007 Jul;21(4):217-31. [ Links ]
70 Min-Oo G, Fortin A, Tam MF, Nantel A, Stevenson MM, Gros P. Pyruvate kinase deficiency in mice protects against malaria. Nat Genet. 2003 Dec;35(4): 357-62. DOI: 10.1038/ng1260 [Link]
71 Ayi K, Min-Oo G, Serghides L, Crockett M, Kirby-Allen M, Quirt I, et al. Pyruvate kinase deficiency and malaria. N Engl J Med. 2008 Apr;358(17):1805-10. [Link]
72 Durand PM, Coetzer TL. Pyruvate kinase deficiency protects against malaria in humans. Haematologica. 2008 Jun;93(6):939-40. [Link]
73 Alves J, Machado P, Silva J, Gonçalves N, Ribeiro L, Faustino P, et al. Analysis of malaria associated genetic traits in Cabo Verde, a melting pot of European and sub Saharan settlers. Blood Cells Mol Dis. 2010 Jan;44(1):62-8. DOI:10.1016/j.bcmd.2009.09.008 [Link]
74 Machado P, Pereira R, Rocha AM, Manco L, Fernandes N, Miranda J, et al. Malaria: looking for selection signatures in the human PKLR gene region. Br J Haematol. 2010 Jun;149(5):775-84. DOI:10.1111/j.1365-2141.2010.08165.x [Link]
75 Souza-Neiras WC, Melo LM, Machado RL. The genetic diversity of Plasmodium vivax: a review. Mem Inst Oswaldo Cruz. 2007 Jun;102(3):245-54. [Link]
76 Langhi DM Jr, Bordin JO. Duffy blood group and malaria. Hematology. 2006 Oct;11(5):389-98. [Link]
77 Tournamille C, Colin Y, Cartron JP, Le Van Kim C. Disruption of a GATA motif in the Duffy gene promoter abolishes erythroid gene expression in Duffy-negative individuals. Nat Genet. 1995 Jun;10(2):224-8. DOI:10.1038/ng0695-224 [Link]
78 Castilho L, Rios M, Pellegrinno J Jr, Saad STO, Costa FF, Reid ME. A novel FY allele in Brazilians. Vox Sang. 2004 Oct;87(3):190-5. DOI:10.1111/j.1423-0410.2004.00554.x [Link]
79 Rowe JA, Opi DH, Williams TN. Blood groups and malaria: fresh insights into pathogenesis and identification of targets for intervention. Curr Opin Hematol. 2009 Nov;16(6):480-7. [Link]
80 Carvalho TL, Ribolla PEM, Curi RA, Mota LS. Characterization and transcriptional analysis of the promoter region of the Duffy blood group, chemokine receptor (DARC) in cattle. Vet Immunol Immunopathol. 2009 Dec;132(2-4):153-9. DOI:10.1016/j.vetimm.2009.05.016 [Link]
81 Allison AC. Genetic control of resistance to human malaria. Curr Opin Immunol. 2009 Oct;21(5):499-505. [Link]
82 Miller LH, Mason SJ, Dvorak JA, McGinniss MH, Rothman IK. Erythrocyte receptors for (Plasmodium knowlesi) malaria: Duffy blood group determinants. Science. 1975 Aug;189(4202):561-3. [Link]
83 Barnwell JW, Nichols ME, Rubinstein P. In vitro evaluation of the role of the Duffy blood group in erythrocyte invasion by Plasmodium vivax. J Exp Med. 1989 May;169(5):1795-802. [Link]
84 Culleton RL, Mita T, Ndounga M, Unger H, Cravo PV, Paganotti GM, et al. Failure to detect Plasmodium vivax in West and Central Africa by PCR species typing. Malar J. 2008 Sep;7:174. DOI:10.1186/1475-2875-7-174 [Link]
85 Kasehagen LJ, Mueller I, Kiniboro B, Bockarie MJ, Reeder JC, Kazura JW, et al. Reduced Plasmodium vivax erythrocyte infection in PNG Duffy-negative heterozygotes. PLoS One. 2007 Mar;2(3):e336. [Link]
86 Ryan JR, Stoute JA, Amon J, Dunton RF, Mtalib R, Koros J, et al. Evidence for transmission of Plasmodium vivax among a duffy antigen negative population in Western Kenya. Am J Trop Med Hyg. 2006 Oct;75(4):575-81. [Link]
87 Cavasini CE, Rossit AR , Couto AA, Couto VS, Gollino Y, Moretti LJ, et al. Duffy blood group gene polymorphisms among malaria vivax patients in four areas of the Brazilian Amazon region. Malar J. 2007 Dec;6:167. DOI:10.1186/1475-2875-6-167 [Link]
88 Cavasini CE, Mattos LC, Couto AA, Bonini-Domingos CR, Valencia SH, Neiras WC, et al. Plasmodium vivax infection among Duffy antigen-negative individuals from the Brazilian Amazon region: an exception? Trans R Soc Trop Med Hyg. 2007 Oct;101(10):1042-4. DOI:10.1016/j.trstmh.2007.04.011 [Link]
89 Anstee DJ. The relationship between blood groups and disease. Blood. 2010 Jun;115(23):4635-43. DOI 10.1182/blood-2010-01-261859. [Link]
90 Rowe JA, Handel IG, Thera MA, Deans AM, Lyke KE, Kone A, et al. Blood group O protects against severe Plasmodium falciparum malaria through the mechanism of reduced rosetting. Proc Natl Acad Sci U S A. 2007 Oct;104(44):17471-6. DOI:10.1073/PNAS.0705390104 [Link]
91 Pant CS, Gupta DK, Sharma RC, Gautam AS, Bhatt RM. Frequency of ABO blood groups, sickle-cell haemoglobin, G-6-PD deficiency and their relation with malaria in scheduled castes and scheduled tribes of Kheda District, Gujarat. Indian J Malariol. 1992 Dec;29(4):235-9. [Link]
92 Beiguelman B, Alves FP, Moura MM, Engracia V, Nunes ACS, Heckmann MIO, et al. The association of genetic markers and malaria infection in the Brazilian western Amazonian region. Mem Inst Oswaldo Cruz. 2003 Jun;98(4):455-60. DOI:10.1590/S0074-02762003000400004 [Link]
93 Migot-Nabias F, Pelleau S, Watier L, Guitard J, Toly C, Ngom MI, et al. Red blood cell polymorphisms in relation to Plasmodium falciparum asymptomatic parasite densities and morbidity in Senegal. Microbes Infect. 2006 Aug;8(9-10):2352-8. DOI:10.1016/J.MICINF.2006.03.021 [Link]
94 Uneke CJ, Ogbu O, Nwojiji V. Potential risk of induced malaria by blood transfusion in South-eastern Nigeria. Mcgill J Med. 2006 Jan;9(1):8-13. [Link]
95 Fischer PR, Boone P. Short report: severe malaria associated with blood group. Am J Trop Med Hyg. 1998 Jan;58(1):122-3. [Link]
96 Lell B, May J, Schmidt-Ott RJ, Lehman LG, Luckner D, Greve B, et al. The role of red blood cell polymorphisms in resistance and susceptibility to malaria. Clin Infect Dis. 1999 Apr;28(4):794-9. [Link]
97 Pathirana SL, Alles HK, Bandara S, Phone-Kyaw M, Perera MK, Wickremasinghe AR, et al. ABO-blood-group types and protection against severe, Plasmodium falciparum malaria. Ann Trop Med Parasitol. 2005 Mar;99(2):119-24. DOI:10.1179/136485905X19946 [Link]
98 Loscertales MP, Brabin BJ. ABO phenotypes and malaria related outcomes in mothers and babies in The Gambia: a role for histo-blood groups in placental malaria? Malar J. 2006 Aug;5:72. DOI:10.1186/1475-2875-5-72 [Link]
99 Uneke CJ. Plasmodium falciparum malaria and ABO blood group: is there any relationship? Parasitol Res. 2007 Mar;100(4):759-65. DOI:10.107/s00436-006-0342-5 [Link]
100 Cserti CM, Dzik WH. The ABO blood group system and Plasmodium falciparum malaria. Blood. 2007 Oct;110(7):2250-8. DOI:10.1182/blood-2007-03-077602. [Link]
101 Loscertales MP, Owens S, O'Donnell J, Bunn J, Bosch-Capblanch X, Brabin BJ. ABO blood group phenotypes and Plasmodium falciparum malaria: unlocking a pivotal mechanism. Adv Parasitol. 2007;65:1-50. DOI:10.1016/S0065-308X(07)65001-5 [Link]
102 Carlson J, Wahlgren M. Plasmodium falciparum erythrocyte rosetting is mediated by promiscuous lectin-like interactions. J Exp Med. 1992 Nov;176(5):1311-7. [Link]
103 Udomsangpetch R, Todd J, Carlson J, Greenwood BM. The effects of hemoglobin genotype and ABO blood group on the formation of rosettes by Plasmodium falciparum-infected red blood cells. Am J Trop Med Hyg. 1993 Feb;48(2):149-53. [Link]
104 Barragan A, Kremsner PG, Wahlgren M, Carlson J. Blood group A antigen is a coreceptor in Plasmodium falciparum rosetting. Infect Immun. 2000 May;68(5):2971-5. [Link]
105 Fry AE, Griffiths MJ, Auburn S, Diakite M, Forton JT, Green A, et al. Common variation in the ABO glycosyltransferase is associated with susceptibility to severe Plasmodium falciparum malaria. Hum Mol Genet. 2008 Feb;17(4):567-76. DOI:10.1093/hmg/ddm331 [Link]
106 van Loon FP, Clemens JD, Sack DA, Rao MR, Ahmed F, Chowdhury S, et al. ABO blood groups and the risk of diarrhea due to enterotoxigenic Escherichia coli. J Infect Dis. 1991 Jun;163(6):1243-6. [Link]
107 Swerdlow DL, Mintz ED, Rodriguez M, Tejada E, Ocampo C, Espejo L, et al. Severe life-threatening cholera associated with blood group O in Peru: implications for the Latin American epidemic. J Infect Dis. 1994 Aug;170(2):468-72.
 Correspondência / Correspondence / Correspondencia:
Correspondência / Correspondence / Correspondencia:
Patrícia Machado
Unidade de Ensino e Investigação de Malária, Instituto de Higiene e
Medicina Tropical
Rua da Junqueira n° 100, 1349-008 Lisboa-Portugal
Tel.:+351 21 365 26 00 (ext. 308)
E-mail: pmachado@ihmt.unl.pt
Recebido em / Received / Recibido en: 27/10/2010
Aceito em / Accepted / Aceito en: 16/12/2010
*Contribuíram igualmente para o presente trabalho.