









 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Curriculum ScienTI
Curriculum ScienTI Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCTION
Malaria remains an important public health problem in subtropical and tropical countries, such as Brazil, where 142,314 people were affected only in 20151,2.The effective treatment of this disease is possible when the diagnosis is early and correct, and Giemsa-stained thick blood smear (GTS) is still the gold standard for this1,2,3,4. Nevertheless, despite its good sensitivity (10-30 parasites/µL), specificity and low-cost, microscopy accuracy may be committed by factors as limited skill of technician, thus PCR-based methods are important alternatives to elucidate diagnosis and for several purposes in research5,6,7,8,9,10,11. The success of PCR depends on purity and integrity of DNA template, which are closely related to extraction method and source of DNA obtainment. Usually, DNA extraction kits present great results, but their high cost leads to the search of in-house protocols, as washing-boiling and the use of DNA chelating agents, like Saponin/Chelex-10012,13,14,15. Regarding to obtainment source, some researches highlighted the filter paper blood spots as the best sources for Plasmodium DNA, since this device allows the simple storage of sample by long-term at room temperature protecting DNA from contamination and degradation, facilitating the sample packing, transportation and manipulation11,12,13,14,15,16.
Other prominent sources are rapid diagnostic tests (RDT) and GTS that show reliable results and wide applicability14,15,16,17,18,19. However, GTS DNA source has additional advantages, because it allows the sample to be used both for microscopy and molecular techniques and can enable retrospective studies from archival blood films, when there is no facility or possibility for blood collection and/or storage. Thereby, in this study it was aimed to evaluate a protocol of nested PCR using GTS as source of Plasmodium DNA from blood films stored until 10 years in a reference laboratory for malaria in endemic area.
MATERIALS AND METHODS
STUDY AREA
This research involved five municipalities from Pará State, Brazilian Amazon Region, where malaria is endemic. The studied localities were: Novo Repartimento (04°14'55''S, 50°07'25''W), Parauapebas (06°04'03"S, 49°54'08"W), Tucuruí (03°45'03"S, 49°40'03"W), located on southeast of Pará State, Ananindeua (01°21'56"S, 48°22'20"W) and Belém (01°27'20"S, 48°30'15"W), located on northeast of Pará State.
ETHICS AND SAMPLE SELECTION
This molecular study to evaluate a protocol of nested PCR using GTS as source of Plasmodium DNA from blood films was approved by the Research Ethics Committee (CEP) of Instituto Evandro Chagas (IEC), Health Surveillance Secretariat (SVS), Brazilian Ministry of Health (MS) - (004/2003-CEP/IEC/SVS/MS).
A total of 138 available GTS slides was used and stored in the Malaria Laboratory at reference center for Brazilian Amazon Region (IEC/SVS/MS). These samples were obtained from inhabitants of the studied municipalities that looked for local health services with suspected malaria between 1997 and 2007. The patients received explanations about the research and its goals, then those who accepted to ingress in the survey answered to clinical-epidemiological records and signed informed consent term. All patients received proper medical assistance.
The sampling was composed by 72 Plasmodium positive GTS (range of parasitaemia: 100-120,000 parasites/µL) and 66 negative (including 25 samples of Trypanosoma cruzi), diagnosed by skilled technicians just after the staining. The slides were classified in three groups: group 1 - 85 GTS (45 positive for Plasmodium and 40 negative) stored in polypropylene box during five years; group 2 - 28 GTS (27 positive for Plasmodium and one negative) stored in wooden box during 10 years; and group 3 - 25 GTS positive for T. cruzi, negative for Plasmodium, stored in polypropylene box during a month. All slides were stored at same room with daily control (twice a day) of temperature (18-25° C) and humidity (average of 75%).
GTS presentation conditions (visual aspect) were evaluated and criteria of smear integrity, oil immersion presence and microbial contamination (presence/absence of bacteria and/or fungi) were observed. Five samples of group 1 were partially unfixed and from group 2, 17 samples were partially unfixed and three of them were also with microbial (bacteria and fungi) contamination. All 138 samples presented rests of immersion oil, but, despite these, all of them were included in this study. As positive controls, filter paper blood spots positives for three species of Plasmodium (mono-infections by Plasmodium vivax, Plasmodium falciparum and Plasmodium malariae) and for T. cruzi were used. Sterile distilled water and non-infected human filter paper blood spots were used as negative controls.
GIEMSA-STAINED THICK BLOOD SMEARS
For each participant, two slides were made and the staining process was performed immediately after the collection and drying of blood samples, as follows: two drops of whole blood (each one with ~ 10 µL of blood) were applied to a clean glass slide to prepare GTS. After drying at room temperature, hemoglobin was removed from GTS by phosphatic methylene blue solution, then, the smears were stained with Giemsa alcoholic solution (5% in buffered solution at pH 7%). Once stained, slides were stored at IEC for reading.
GTS were examined at 1,000x to identify parasite species and determine the parasite density, and it was calculated by counting the number of asexual parasites using a hand tally counter against 200 white blood cells (WBC), based on a WBC count of 6,000 WBC/µL. All blood smears were examined by two independent microscopists at IEC. They reviewed each other's results on a blinded way. Blood smears with differences in species diagnosis or asexual parasite density of > 50% difference in asexual parasite density between the two microscopists would be re-examined by a third independent microscopist also at IEC20.
EXTRACTION, QUANTIFICATION AND QUALITY EVALUATION OF DNA
This protocol was performed according to Alger et al14. Firstly, all slides were washed with saline solution (pH 7.0), afterwards each smear was scraped of the slide surface and mixed with 500 µL of Saponin aqueous solution (1%) in a 1.5 mL microtube. Then, the samples were incubated on ice for 1 h, vortexed 3-4 times during this time and then centrifuged for 4 min at 12,000x g at room temperature. The supernatants were discarded and the pellets were washed (2-3 times) in periplural blood smear (PBS) (pH 7.2), mixed to 50 µL of Chelex-100 aqueous suspension (5%) and incubated at 100° C on dry heating block for 20 min. After that, the samples were centrifuged at 12,000x g for 4 min and the supernatants were transferred to new microtubes, following the quantification of samples and evaluation of purity and integrity. Main steps of this protocol are shown in figure 1.

Positive and negative controls (filter paper blood spots) were similarly extracted. It was used 1/4 of the blood spot that was cut in small pieces and incubated with Saponin solution 0.5% in PBS and in ice bath for 1 h. After, the solution was vortexed and the supernatant was discarded. Then, a wash with PBS was conducted followed by addition of 50 µL of Chelex-100 aqueous suspension (20%) and incubation on dry heating block at 100° C for 1 h. Finally, the material was centrifuged and the supernatant was transferred to new microtubes and kept at -20° C according to Wooden et al13.
The DNA quantification was realized by spectrophotometry (GeneQuantTM II, Pharmacia Biotech, Piscataway, New Jersey, USA) with determination of A260/A280 (accepted range for DNA template: 1.5-2.0 ng/mL). For DNA structural integrity evaluation, DNA samples and DNA ladder (100 pb) were fragmented by electrophoresis using 1% agarose gel stained with ethidium bromide (5 µL/mL in Tris-Borate-EDTA buffer) for 20 min and visualized with ultraviolet light (320 mn).
NESTED PCR
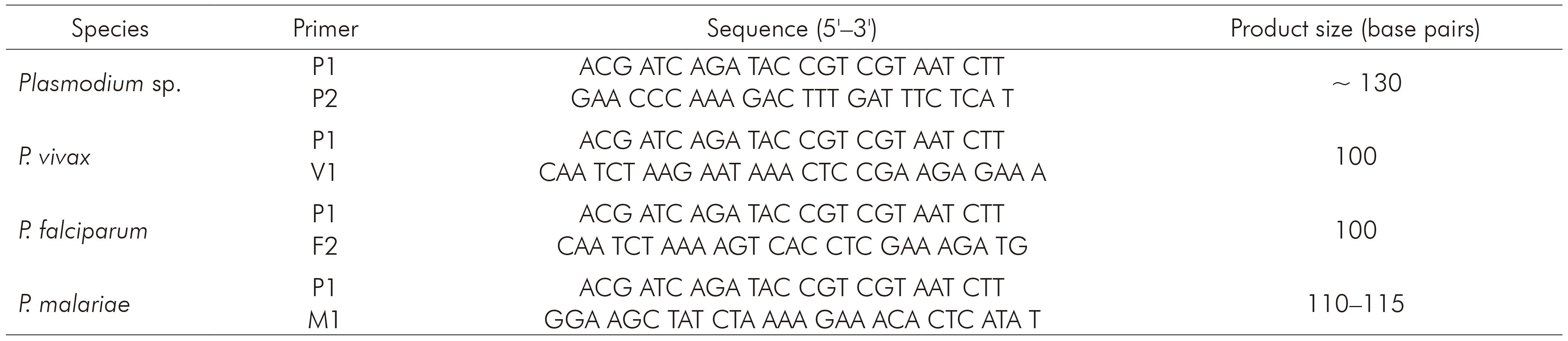
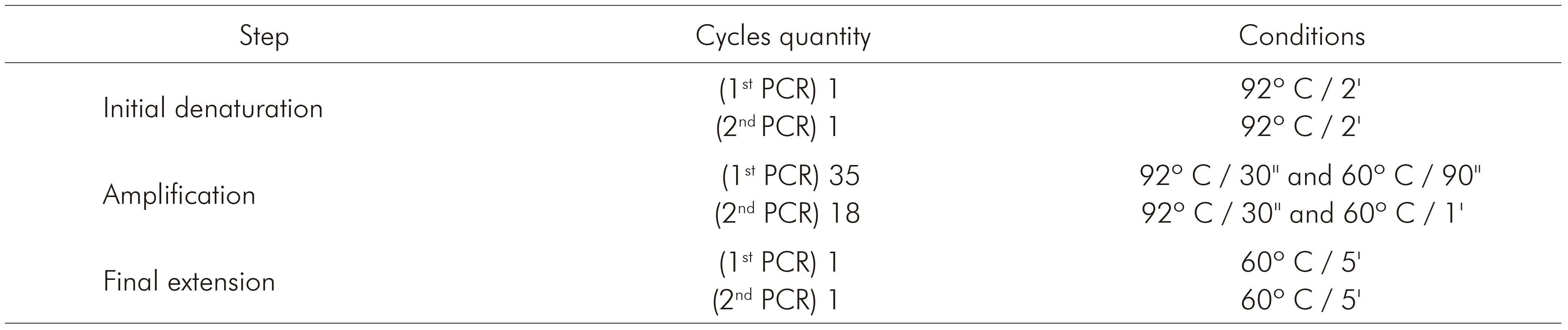
The nested PCR for detecting the three species of Plasmodium that occur in Brazil (P. vivax, P. falciparum and P. malariae) was processed using primers described by Kimura and others7, whose protocol is based on amplification of 18S rRNA. Primers and products sizes are described in table 1. Briefly, 5 µL of extracted DNA were used to amplify genus-specific fragment with primers P1 and P2. The final volume of this reaction was 20 µL, as follows: 0.4 µL of P1 and P2, 312.5 µM of each dNTP, buffer (50 mM KCl, 10 mM Tris-HCl and pH 8.3), 1.5 mM MgCl2 and 0.75 U of Taq DNA polymerase. For the amplification of the specie-specific fragments, 2 µL of the product from the first reaction (1:40) were used with 1 µM of P1 and 1 µM of V1, F2 or M1, 312.5 µM of each dNTP, buffer (50 mM KCl, 10mM Tris-HCl and pH 8.3), 1.5 mM MgCl2 and 0.75 U of Taq DNA polymerase. Amplification programs are described in table 2. PCR products were fragmented by 2% agarose gel electrophoresis and stained with ethidium bromide (5 µL/mL in Tris-Borate-EDTA buffer).
STATISTICAL ANALYSIS
Screening test was used in order to determine sensitivity, specificity, predictive value of positive test, predictive value of negative test, false-positive, false-negative and accuracy. Adopted significance level was 0.0521.
RESULTS
DNA from positive GTS of group 1 showed concentrations ranging between 17 ng/µL and 119 ng/µL (average = 66.7 ng/μL ± 31.5 ng/μL) and from negative GTS between 10 ng/µL and 19 ng/µL (average = 13.65 ng/μL ± 3.13 ng/μL). In group 2, DNA samples from GTS Plasmodium positive ranged between 14 ng/µL and 90 ng/µL (average = 48.30 ng/μL ± 28.41 ng/μL) and the only GTS negative had 10 ng/µL. T. cruzi samples of group 3 ranged between 15 ng/µL and 21 ng/µL (average = 17.48 ng/μL ± 2.08 ng/μL).
The ratio A260/A280 nm of positive samples from group 1 ranged between 1.75 and 1.86 (average = 1.8 ± 0.04) and of negative samples ranged between 1.61 and 1.82 (average = 1.75 ± 0.07). Positive samples from group 2 ranged between 1.65 and 1.84 (average = 1.76 ± 0.06) and the negative sample showed 1.62. Samples from group 3 ranged between 1.68 and 1.82 (average = 1.76 ± 0.05). The evaluation of DNA integrity in agarose gel showed high molecular weight bands to all samples of group 1, while in group 2, 19 did not present any band. Figure 2 shows different nested PCR profiles of integrated and degraded samples of the three groups. Statistical parameters for Plasmodium detection by nested PCR in comparison to microscopy are shown in table 3. Regarding to group 3, all samples were negative for Plasmodium, which were confirmed by nested PCR, eliminating the possibility of cross reaction with T. cruzi. The comparison between microscopy and nested PCR are shown in table 4.
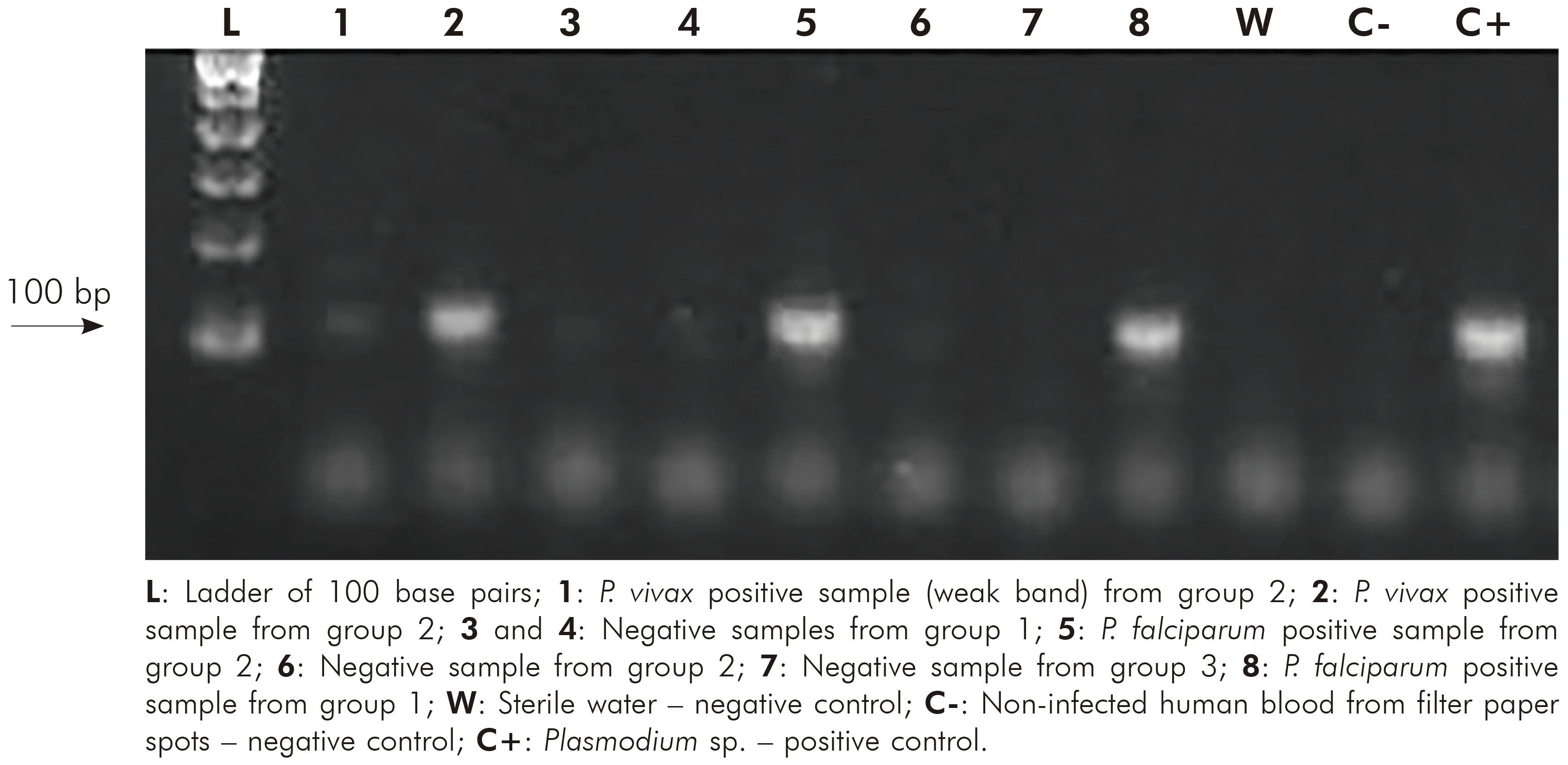
Table 3 - Statistical parameters calculated for Plasmodium detection by nested PCR in comparison to microscopy
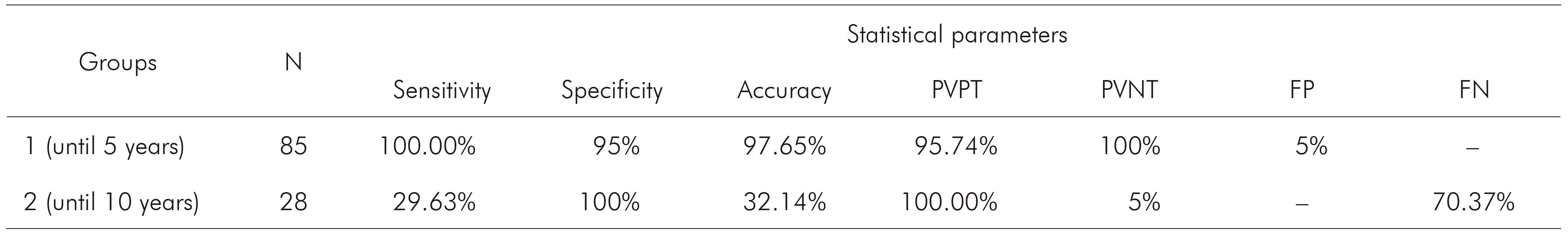
N: Number of samples per group; PVPT: Predictive value of positive test; PVNT: Predictive value of negative test; FP: False-positive; FN: False-negative; Conventional sign used: - Numerical data not equal to zero due to rounding.
Table 4 - Comparison between results of microscopy and nested PCR in each group

Pf: P. falciparum; Pv: P. vivax, Pm: P. malariae; Pf + Pv: Mixed infection by P. falciparum and P. vivax; Pf + Pm: Mixed infection by P. falciparum and P. malariae; Conventional sign used: - Numerical data not equal to zero due to rounding.
DISCUSSION
Gold standard microscopy may not answer many scientific and routine issues related to malaria that many times can be solved by molecular analysis, mainly due to the parasites identification in areas where Plasmodium species occur simultaneously. To perform PCR protocols, it is necessary high quality DNA template that depends on factors as its source. Factors as DNA amount, high content of haemoglobin or human DNA, use of anticoagulant as heparin, inadequate conditions of blood collecting, storage and amplification of samples may inhibit the PCR assay6,8. Meanwhile, one of the major problems to perform PCR in malaria researches is the limitation of DNA sources16, making it difficult to conduct retrospective studies, which are fundamental to comprehension of parasite features (like resistance traits) and population genetics. Thus, the use of alternative sources of nucleic acid is very important and has several applications in health surveillance.
The storage of blood samples with anticoagulant or in filter paper increases costs and usually is restrict to reference centers. Moreover, new collections of blood samples may be difficult due to the impossibility to find out or to reach patients, especially in remote areas in the Amazon Region. This region has large territorial extension (4,871,500 km², corresponding to 57.3% of Brazilian territory) and logistical challenges of access to some malaria endemic areas. This retrospective study has evaluated the sensitivity and specificity of PCR to detect malaria parasites in accordance of the blood conservation in GTS device used for DNA extraction. The use of DNA from GTS had demonstrated that Plasmodium DNA might be successfully isolated from this device indicating that this method of DNA preservation could be considered viable and convenient for epidemiological studies6,14. Thus, this study attended the malaria researches challenging according with lack of samples to diagnostic clarification and decrease costs.
Two groups (1 and 2) of samples were included in order to investigate the viability of Plasmodium DNA obtained from GTS stored for five and 10 years, respectively. It was also added a third group (group 3) with T. cruzi (another hemoparasite endemic in studied area) samples to verify specificity and possibility of cross reaction, which was not observed. Regarding the group 1, the results showed sensitivity of 100% and specificity of 95% by nested PCR. Furthermore, nested PCR detected two mixed infections that were not identified by microscopy. Whether these two individuals with negative GTS but positive nested PCR may act as reservoirs of the parasite, it remains unclear. However, it is known that in endemic area it is most probable that the PCR actually detects the infection. To support this assumption, it is advisable to conduct a prospective study in the symptomless individuals to confirm the infection status. Beside to this, the values of DNA quantification, purity and integrity showed that the GTS samples stored until five years are convenient source to perform DNA isolation and nested PCR.
Several studies have supported GTS as practicable alternative source of Plasmodium DNA6,14,15,17. In this survey, in spite of some results that showed switch of Plasmodium species by nested PCR, this inference is also possible, since these differences occurred due limitations of GTS reading protocol adopted in Brazil, similar to other study15. Here, 14 mono-infection samples by microscopy were mixed infection by nested PCR, two P. falciparum samples by microscopy were P. vivax by molecular protocol and two mixed infections by microscopy were mono-infections by nested PCR and all of them presented immature forms parasites that can confuse even skilled microscopists.
According to technical note adopted in Brazil22, P. falciparum + P. vivax mixed infections shall be registered in mid or high-level parasitaemia of P. falciparum young trophozoites (ring forms) without Shüffner granulations associated with mature trophozoites (irregular forms) and/or gametocites with Shüffner granulations of P. vivax in lower parasitaemia level, even in the absence of typical P. falciparum gametocites. P. falciparum mid or high-level parasitaemias only with ring forms (independent of evolutive forms) is registered as mono-infection23.Meanwhile, the presence of only trophozoites in several evolutive forms at erythrocytic schizogony was already reported as suggestive of mixed infection, because at that moment typical forms of each specie have not yet reached peripheral circulation, ensuring accuracy of mixed infection8,24.
The current study did not aim to survey mixed infection, but it was suggested that countries where malaria is endemic must consider this and perform studies to review their protocols to precise identification of mixed infection by microscopy. Beside this, they shall consider to adopt PCR based-techniques to identify the real prevalence of Plasmodium species contributing to disease epidemiology, correct malaria therapy and, consequently, to World Health Organization strategies to guarantee malaria surveillance into a core intervention and to accelerate efforts towards elimination and attainment of malaria-free status4,25.
Regarding to samples stored for 10 years (group 2), the study showed that they were not reliable to Plasmodium molecular detection. Some GTS were contaminated, unfixed and presented degraded parasites, which compromised DNA quality. It was not verify the humidity inside the boxes where the slides were stored, but it was supposed that, beyond the long time of storage, these samples were also affected due the storage conditions in wooden box, which contributed to the microorganism proliferation. The evaluation of DNA quantity and purity indicated acceptable values to perform nested PCR, but DNA isolated from 19 samples of this group were degraded, resulting in false-negative results by molecular assay and, therefore, low sensitivity and accuracy. Factors involved in preparing slides for microscopic assay may contribute to the stability of DNA template. Furthermore, Giemsa-stain step and excessive manipulation of the slides will be able to act negatively on DNA integrity8,14.
Other study also observed low sensitivity of PCR using DNA from GTS, but, despite this, the authors concluded that DNA obtained from GTS can be used in nested PCR to amplify 18 SSU rRNA gene of Plasmodium species in retrospective surveys, empathizing the limitation of this in low-level parasitaemias7. The current survey did not observe the relation between parasitic density and nested PCR sensitivity. To this purpose, the samples should be increased and make further studies. Not with standing, this study was able to demonstrate that GTS samples stored in controlled conditions of humidity and temperature until five years are a consistent alternative to obtain Plasmodium DNA, even using in-house protocol of DNA extraction. The assumption is that even older samples can be useful as DNA source if guaranteed good storage conditions26.
The choice of extraction methods based on in-house protocol used to be cheaper than commercial kits. In this study, Saponin/Chelex-100 for DNA extraction showed similar performance as other commercial protocols16,27. Thus, the findings highlight that it is possible to implement simple DNA extraction protocol from GTS that can be used as historical DNA source for several aims, drug resistance studies as for example. However, proper storage conditions are required to guarantee the integrity of the genomic DNA used for analysis. Attempts to evaluate simple molecular tools should continue, especially in settings where is difficult to obtain samples and to perform retrospective studies.
CONCLUSION
In this survey, it was observed that in-house protocol of DNA extraction using Saponin/Chelex-100 is able to isolate Plasmodium DNA from GTS samples stored for five years. Otherwise, GTS samples stored for 10 years were not reliable as Plasmodium DNA source, probably due the storage conditions. Finally, it was concluded that nested PCR for Plasmodium detection using DNA from archival GTS stored up to five years is a very useful protocol for malaria diagnosis, being especially important in retrospective studies and when another sample collection is not feasible.