









 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Curriculum ScienTI
Curriculum ScienTI Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCTION
Recommended in a variety of situations, the analysis of the chromosomal constitution of patients remains one of the most commonly performed genetic tests1,2. Chromosomal abnormalities are associated with more than 60 identifiable syndromes and rank among the chief causes of intellectual disability and congenital malformations, including facial dimorphism, skeletal abnormalities, congenital heart defects, and abnormal sex development3,4,5. Moreover, they can even be involved in the pathogenesis of some cancers3.
In terms of classification, chromosomal abnormalities can be divided into numerical and structural abnormalities, the latter of which are either balanced or imbalanced. Among numerical changes are aneuploidies, which are characterized by the presence or absence of one or more chromosomes of certain pairs in individual cells. By contrast, cases of loss, gain, and inversions of chromosome segments, as well as the translocation of segments to different chromosomes, are examples of structural rearrangements. Both, numerical and structural changes can be identified in a state of mosaicism, in which only a portion of the cells forming patient's body is affected by the mutation. The degree of mosaicism is often reflected in the level of aggressiveness of a disease1.
As G-banding analyses have revealed, chromosomal abnormalities are estimated to be present in at least 50.0% of spontaneous abortions, and approximately 0.5% of newborns are affected by genetic alterations4, of which 0.3% are numerical changes3. The mother's age is considered to be a risk factor of the development of chromosomal abnormalities in uterus, and women aged 35 years or older currently account for about 2.0% of all pregnancies with such abnormalities3,4.
In Brazil, most care facilities offering genetic counseling and examinations are concentrated in Southeastern and Southern Brazil and are usually integrated with universities and reference hospitals5. By comparison, few facilities offering such services are found elsewhere in the country. For instance, in Pará State, Northern Brazil, cytogenetic diagnostic tests are offered by only one laboratory of Universidade Federal do Pará (UFPA). This laboratory (Human and Medical Cytogenetics Laboratory) has offered cytogenetic examinations for patients at the Hospital Universitário Bettina Ferro de Souza (HUBFS) since 1997.
Although such examinations are fully financed by the Brazilian Unified Health System (Sistema Único de Saúde - SUS), like many services in Brazil, the laboratories which offer them lack for organization in a network and are not officially integrated into the national health system, despite the great importance of the records that they produce5,6. Given the absence of records with epidemiological data representing chromosomal abnormalities in Brazilian Amazon Region, the aim of this study was to analyze the frequency and types of chromosomal abnormalities in patients referred to the Human and Clinical Cytogenetics Laboratory (HCCL) in Belém, Pará State. The data represent a 17-year period and relate some of the chromosomal findings to clinical records, patient age, and other available medical records.
MATERIALS AND METHODS
This study was based on a retrospective survey conducted from 1997 to 2014 and was approved by UFPA Institutional Ethics Research Committee (652.977/2014, 05/20/2014). Analyses were based on information registered during patient evaluation, including clinical findings, age, possible consanguinity, and other cases of family abnormalities, as well as chromosome G-banding results.
The sample consisted of 1,580 patients referred by the pediatric service of HUBFS in Belém. Cytogenetic analyses were performed on cultured peripheral blood lymphocytes stimulated with phytohemagglutinin, and karyotypes were determined by G-banding7,8.
Clinical data were grouped according to the International Classification of Diseases, 10th edition (ICD-10)9, and a frequency survey of each group was performed. The clinical findings mentioned in Chapter XVII regarding congenital malformations, deformations, and chromosomal abnormalities were subgrouped according to shared clinical features for more detailed analysis. Afterward, the frequency of abnormal karyotypes for each group and subgroup was evaluated.
Statistical analysis considered the affected chromosome pair, the type of chromosomal change (i.e., numerical or structural), and the type of chromosome affected (i.e., autosomal or sex). Data were analyzed by Mann-Whitney test (GraphPad Prism v5.0).
RESULTS
Participants' age ranged from 2 days old to 36 years old ( = 11.5 ± 10.6 years), from a total of 1,580 patients, of which 730 presented chromosomal abnormalities. Frequently, participants with sex chromosome abnormalities (N = 206) sought medical care at an older age than those with autosomal abnormalities (N = 524) (Figure 1), with peaks around 11-15 years old (30.1% of cases) and 0-6 months old (40.5%), respectively. By contrast, participants with normal karyotypes (N = 850) peaked at an intermediate age (i.e., 4-10 years old), and the means of the three groups were statistically different (p < 0.0001).
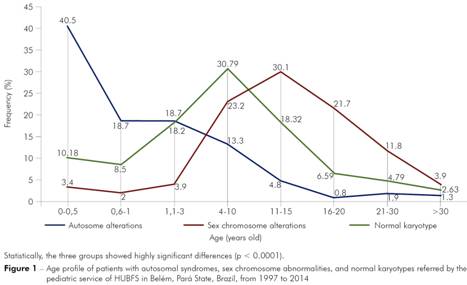
Statistically, the three groups showed highly significant differences (p < 0.0001).
Figure 1 - Age profile of patients with autosomal syndromes, sex chromosome abnormalities, and normal karyotypes referred by the pediatric service of HUBFS in Belém, Pará State, Brazil, from 1997 to 2014
Of the 1,580 participants, 1,392 (88.1%) presented clinical features or syndromic profiles identified in Chapter XVII of the ICD-10 related to congenital malformations, deformities, and chromosomal abnormalities. The distribution of clinical features among participants according to the affected body system indicated in the ICD-10 is shown in table 1. Suspected participants of the most common chromosomal syndromes, including Down, Klinefelter, and Turner, represented 53.0% of cases. Other participants had congenital malformations not associated with a particular syndrome; those involving eyes, ears, face, and neck (Q10-Q18) were the most frequent (14.6%), followed by ones in the sexual organs (Q50-Q56, 12.3%), and ones in the musculoskeletal system (Q65-Q79, 7.0%).
Table 1 - Total and absolute frequency of congenital malformations, deformations, and chromosomal abnormalities pattern in ICD-10 classification9 in patients enrolled in HCCL/UFPA, from 1997 to 2014
| Chapter XVII: Congenital malformations, deformations and chromosomal abnormalities | |||
|---|---|---|---|
| Blocks | Title | Total cases | % |
| Q00-Q07 | Congenital malformations of the nervous system | 44 | 3.2 |
| Q10-Q18 | Congenital malformations of eye, ear, face and neck | 203 | 14.6 |
| Q20-Q28 | Congenital malformations of the circulatory system | 22 | 1.6 |
| Q30-Q34 | Congenital malformations of the respiratory system | - | - |
| Q35-Q37 | Cleft lip and cleft palate | 22 | 1.6 |
| Q38-Q45 | Other congenital malformations of the digestive system | 6 | 0.4 |
| Q50-Q56 | Congenital malformations of genital organs | 171 | 12.3 |
| Q60-Q64 | Congenital malformations of the urinary system | 3 | 0.2 |
| Q65-Q79 | Congenital malformations and deformations of the musculoskeletal system | 98 | 7.0 |
| Q80-Q89 | Other congenital malformations | 37 | 2.6 |
| Q90-Q99* | Chromosomal abnormalities, not elsewhere classified | 737 | 53.0 |
| Anomaly unspecified | 49 | 3.5 | |
| Total | 1,392 | 100.0 | |
* All cases of patients with specific syndromic profile were considered, regardless of the karyotype result; Conventional sign used: - Numerical data not equal to zero due to rounding.
Mental and behavioral disorders (F00-F99) without syndromic features were the second-most frequent (18.1%). Most of the participants in that group showed delays in neuropsychomotor development, as well as learning and language disorders. Endocrinal, nutritional, and metabolic disorders (E00-E90) were detected in 7.2% of participants, most of them exhibited small stature (Table 2).
Table 2 - Frequency of clinical findings in patients analyzed, grouped into securities, according to the ICD-10
| Chapter | Blocks | Title | Total cases | % |
|---|---|---|---|---|
| II | C00-D48 | Neoplasms | 3 | 0.2 |
| III | D50-D89 | Diseases of the blood and blood-forming organs and certain disorders involving the immune mechanism | 4 | 0.2 |
| IV | E00-E90 | Endocrine, nutritional and metabolic diseases | 147 | 7.2 |
| V | F00-F99 | Mental and behavioural disorders | 369 | 18.1 |
| VI | G00-G99 | Diseases of the nervous system | 11 | 0.5 |
| VII | H00-H59 | Diseases of the eye and adnexa | 11 | 0.5 |
| VIII | H60-H95 | Diseases of the ear and mastoid process | 8 | 0.4 |
| X | J00-J99 | Diseases of the respiratory system | 1 | 0.1 |
| XI | K00-K93 | Diseases of the digestive system | 27 | 1.3 |
| XII | L00-L99 | Diseases of the skin and subcutaneous tissue | 7 | 0.3 |
| XIV | N00-N99 | Diseases of the genitourinary system | 43 | 2.1 |
| XVI | P00-P96 | Certain conditions originating in the perinatal period | 10 | 0.5 |
| XVII | Q00-Q99 | Congenital malformations, deformations and chromosomal abnormalities | 1,392 | 68.4 |
| XVIII | R00-R99 | Symptoms, signs and abnormal clinical and laboratory findings, not elsewhere classified | 3 | 0.2 |
| Total | 2,036 | 100.0 |
Mental and behavioral disorders were associated with all participants with congenital anomalies in the nervous system, eyes, ears, face, and neck; whereas metabolic, nutritional, and endocrinal disorders were more frequent in participants with malformations in the sexual organs.
CYTOGENETIC DIAGNOSTICS
Results for chromosomal abnormalities, which were identified in 730 participants (46.2%), appear in table 3. Numerical alterations were the most frequent, being detected in 637 (87.3%), whereas structural chromosomal alterations accounted for 93 cases (12.7%). Ten patients (1.4%) presented both numerical and structural aberrations. Mosaicism was observed in 60/637 (9.4%) of patients with numerical alterations and in 18/93 (19.3%) of patients with structural alterations. Moreover, all cases with both numerical and structural chromosome alterations (10 cases, 1.4%) exhibited mosaicism. A total of 524 (71.8%) abnormalities involved autosomes, while sex chromosomes were involved in alterations of 206 (28.2%) patients.
Table 3 - Absolute and relative frequency of the main syndromes/chromosomal alterations. The results were divided between changes in autosomes and sex chromosomes
| Autosomal chromosome | ||||
|---|---|---|---|---|
| Syndrome | Total cases | % between autosomal | % of total cases | Specific changes |
| Down | 424 | 80.9 | 58.1 | Trisomy: 406 Translocation: 18 |
| Cri-du-chat | 7 | 1.3 | 1.0 | Deletion 5p-:7 |
| Edwards | 6 | 1.2 | 0.8 | Trisomy: 5 46,XX,i(18q): 1 |
| Patau | 2 | 0.4 | 0.3 | Trisomy 13: 2 |
| Other changes | 85 | 16.2 | 11.6 | Deletion: 39 Duplication: 22 Translocation: 11 Inversion: 8 Isochromosome: 2 Marker chromosome: 2 Derivative chromosome: 1 |
| Total | 524 | 100.0 | 71.8 | |
| Sex chromosome | ||||
| Syndrome | Total cases | % between sex chromosomes | % of total cases | Specific changes |
| Turner | 175 | 84.9 | 24.0 | 45,X: 59 45,X/46,XX: 42 Partial deletion of chromosome X: 25 Partial deletion of chromosome X in mosaicism: 18 46,XX,i(Xq ou Xp): 15 45,X/ Partial deletion of chromosome X: 7 45,X/46,Xi(Xq): 2 45,X/ 46,XY: 3 45,X/46,XX/47,XXX ou XYY: 3 45,X/46,X,r(X)/46,XY: 1 |
| Klinefelter | 25 | 12.1 | 3.4 | 47,XXY: 12 47,XXY;46,XY: 8 48,XXXY: 2 46,XY/47,XXY/48,XXXY/49,XXXXY: 3 |
| 47,XXY | 3 | 1.5 | 0.4 | Mosaicism: 1 |
| 48,XXXX | 1 | 0.5 | 0.1 | |
| 47,XXY/46,XY/46,XX | 1 | 0.5 | 0.1 | |
| Other changes | 1 | 0.5 | 0.1 | Duplication: 1 |
| Total | 206 | 100.0 | 28.2 | |
Down syndrome was the most frequently found chromosomal abnormality, observed in 424/730 (58.1%), and the typical karyotype (47,XX+21 or 47,XY+21) was observed in 406/424 (95.8%). Most participants with Down syndrome were male (57.2%). The analysis of the medical records of those participants revealed the recurrence of Down syndrome among relatives in 72/424 cases (17.0%), and seven (1.7%) of the cases had consanguineous parents.
Turner syndrome was detected in 175/730 patients (24.0%). Among them, only 59/175 (33.7%) exhibited the pure classical karyotype of the syndrome (45,X). Mosaicism was identified in 76/175 (43.4%) participants with Turner syndrome, and 58/175 (33.1%) of them showed only X-chromosome structural alterations, either pure or in mosaicism.
Among structural aberrations, deletion was the most frequent abnormality. Among autosomal abnormalities, deletions were observed in 46/524 (8.8%) patients, and pairs 1, 3, 5, 9, and 16 were the most affected. Duplications were found in 22/524 patients, mostly affecting pair 16 (11/22; 50.0%) and 9 (5/22; 22.7%). Additionally, pair 9 was involved in the only case of isochromosome observed and was the most frequently altered by inversion.
DISCUSSION
As shown in table 4, which compares the results of the present study with the findings of other studies in the Brazilian population, this was the first to investigate the occurrence of chromosomal abnormalities in a sample from Northern Brazil.
Table 4 - Comparison of the findings collected in this study and previously reports from Brazil
| Present study | (10) | (11) | (12) | (13) | (14) | (15) | (16) | |
|---|---|---|---|---|---|---|---|---|
| State | Pará | Rio de Janeiro | Rio Grande do Sul | São Paulo | São Paulo | Mato Grosso | São Paulo | Paraná |
| Type of service | Clinical cytogenetics service | Clinical genetics service | Genetic counseling | Ambulatory | Ambulatory | Clinical cytogenetics service | Ambulatory | Clinical genetics service |
| Period | 1997-2014 | 2 years | 1986-2002 | 1998-1999 | 1992-2002 | 2003-2007 | 2006-2009 | 2011-2012 |
| Number of patients analyzed | 1,580 | 98 | 916 | 1,091 | 1,122 | 585 | 317 | 202 |
| Number of analyzed metaphases | 30, toM:50 | 30, toM:100 | 30, toM:100 | - | - | 20, toM:30* | - | - |
| % of abnormal cases | 46.2 | 26 | 29.5 | 23.0 | 22.0 | 20.0 | 10.7 | 15.8 |
| % of numerical changes | 75.3 | 82 | 56.3 | - | 70.8 | 79.5 | 91.2 | 71.9 |
| % Down | 58.1 | 42.9 | 40.5 | 32.0 | 47.4 | 53.9 | 52.9 | 46.9 |
| % Edward | 0.8 | 7.1 | 2.6 | 12.0 | 7.3 | 5.1 | 8.8 | 0.0 |
| % Patau | 0.3 | 3.6 | 1.5 | 3.6 | 0.9 | 2.9 | 0.0 | |
| % Cri-du-chat | 1.0 | 3.6 | 1.1 | 4.0 | 3.6 | 2.6 | - | 0.0 |
| % Turner | 24.0 | 17.9 | 10.8 | 6.0 | 7.3 | 13.7 | 11.8 | 12.5 |
| % Klinefelter | 3.4 | 7.1 | 1.9 | 1.3 | 0.4 | 2.6 | 11.8 | 6.3 |
| % others | 12.5 | 17.9 | 41.6 | 44.7 | 30.4 | 21.4 | 11.8 | 34.3 |
* toM = to mosaicism.
Although other studies have reported a frequency of abnormal karyotypes in 10.7-29.5% of patients10,11,12,13,14,15,16, the frequency showed in this study was far higher (45.6%, 720/1,580 cases). The difference could stem from the eligibility criteria used to refer patients for cytogenetic analyses; 88.1% of patients (1,392/1,580) had some congenital malformation, while 837/1,580 (53.0%) were suspected of having a specific syndrome. Aside from those values, the types and frequencies of abnormal karyotypes were quite similar to the results of previous studies in the Brazilian population. These data agree with those studies, as there was also a predominance of numerical alterations versus structural chromosomal abnormalities, namely in a ratio of 3:1. Duarte et al.11 and Vasconcelos et al.13 also compared the frequencies of autosomal and sex chromosome alterations and found results (83.7%, 16.4%, 86.2%, and 10.5%, respectively) highly similar to the present study: 71.8% for autosomes and 28.2% for sex chromosomes.
The same situation arose regarding the most common chromosomal syndromes. As in other studies, Down syndrome was the most frequent among patients (58.1%). For instance, Duarte et al.11 reported a group profile of patients diagnosed with Down syndrome highly similar to this study: 93.6% of cases with trisomy 21 and a slight predominance of males (54.6%). However, Patau syndrome and Edwards syndrome represented only 1.1% of cases, whereas in previous studies they were detected at a frequency of 10.9% to 12.0%12,13,15. The low value could be associated with the poor quality of neonatal public treatment in Northern Brazil, which, together with Northeast Region, presented the most serious structure problems, especially in public hospitals17.
Karyotypes with structural abnormalities were identified in 21.19% of all cases involving autosomal chromosomes (111 of 524). In particular, translocations involving pair 21 were found in 18 patients, whereas 5p deletions were observed in seven patients and an isochromosome 18q in one case. Two cases exhibited 16q11 duplication. The clinical characteristics of patients in such cases included functional alterations with global disorders, behavior disturbances, and learning difficulties, as well as anatomic alterations such as low-set ears, facial alterations, and extremities malformations. According to previous research, 16q11.2 microamplifications and microdeletions have been associated with cognitive and behavioral alterations and ranked among the most common genetic causes of susceptibility to autism spectrum disorders18.
Regarding sex chromosomes, Turner syndrome was the most frequently observed in sample (84.94%), at a rate seven times greater than Klinefelter syndrome. Interestingly, Klinefelter syndrome has been reported in approximately 1:600-1,000 of live males born19, whereas Turner syndrome has been reported in only 1:3,000-5,000 live girls born20,21. That difference and similar ones observed by other specialists could stem from the syndromes phenotypes10,11,12,14,16, as phenotypic alterations of Turner syndrome are readily observable, whereas those of Klinefelter syndrome are only observable in puberty. Furthermore, these results suggest that a high proportion of individuals with Klinefelter syndrome have not been diagnosed22.
In recent decades, Brazil has experienced an epidemiological transition in which birth defects and genetic diseases have begun to play a more important role in morbidity and mortality rates, especially at reference hospitals5,23. According to Brazilian government records, 33,044 admissions for birth defects and chromosomal diseases, in addition to 32,173 for mental disorders, were registered in public hospitals in Pará State during 1997-201424. Those figures suggest that the total number of individuals referred for cytogenetic diagnostic examination for both type of condition in HCCL/UFPA corresponded to approximately 4% and 1% of the population, respectively. The numbers also clearly indicate the limited access of the Brazilian population to examinations, which is exacerbated by the fact that HCCL/UFPA is the only SUS laboratory in Pará to offer such services. Reflecting the dearth of facilities, as well as the lack of public investment in them, the long waiting times for examinations also pose another major impediment for the Brazilian population5.
Given the lack of knowledge among pediatricians and other health professionals, chromosomal diseases in Brazil are diagnosed late, or else patients are not referred to genetic services5,22, both of which are especially true regarding sex chromosome abnormalities. In this study sample, more than 51% of patients with sex chromosome alterations were referred to cytogenetic services at 11-20 years of age (Figure 1). However, early diagnosis of sex chromosome syndromes may provide better development and quality of life for patients with these conditions. As Linglart et al.25 observed, hormone reposition in patients with Turner syndrome may reduce likelihood of developing short stature. Similarly, Samango-Sprouse et al.26 observed significant advantages in the cognition, language, intellectual, and psychomotor functions of patients with Klinefelter syndrome treated with hormone therapy at 3-6 years old compared to a group of untreated patients.
It is clear that implementing and improving genetic counseling services are of urgent in Brazil and other developing countries, as Lee and Thong27 have previously proposed. These actions aim to help people understand and adapt to the medical, psychological, and familial consequences of the genetic diagnosis of a clinical condition28.
CONCLUSION
The cytogenetic evaluation of patients with congenital abnormalities or functional disorders, especially neural, endocrine, and metabolic, represents an important step in the process of diagnosing specific conditions. Nevertheless, some Brazilian regions, especially Northern, lack systems to diagnose and monitor chromosomal abnormalities, although most of them are fast, specific, and relatively inexpensive, even on a large scale. The adoption of certain public actions designed to benefit the diagnosis, care, and early treatment of patients with chromosomal conditions may help in the development and quality of life. Moreover, surveys like this may serve as guidelines for government projects focused on chromosomopathies affecting the population. Finally, while the G-banding technique remains the gold standard for all cytogenetic techniques, it detects translocations, inversions, deletions, and insertions involving segments of only 7-10 Mb at least. The use of more refined methods such as fluorescence in situ hybridization and array-based techniques, however, could increase the occurrence of structural rearrangements, especially microdeletions and microduplications.