









 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCTION
Brazilian chicken meat production grew 4.5% in 2020 compared with the previous year, reaching the total volume of 13,845 million metric tons. It was exported 4,231 million metric tons, generating revenues of US$ 6,097 million; Brazil ranked among the largest producers, the third after the United States and China but it was the world leader in exports of this protein1.
The poultry production in Pará State is growing each year and represents the second most prominent activity of the agricultural sector in the state2. However, numerous reports reveal the circulation of viral pathogens in the global production of broiler chickens, contributing to significant economic losses3,4.
Caliciviruses (CV), belonging to the Caliciviridae family, are currently classified into 11 genera: Bavovirus, Lagovirus, Minovirus, Nacovirus, Nebovirus, Norovirus, Recovirus, Salovirus, Sapovirus, Valovirus, and Vesivirus5. They are single-stranded RNA viruses with positive-sense, non-segmented, and polyadenylated genomes of about 6.4 to 8.5 kilobases (kb). A non-enveloped icosahedral capsid encapsulates their genome with 27-42 nm in diameter. The genomic structure is variable according to the genus and contains in its genomic extension two to four open reading frames (ORF) covalently linked to the viral protein genome-linked (VPg) at the 5' end and a poly (A) tail of variable length at the 3' end6.
In this study, we present the genomic characterization of a Bavovirus, a genus recently included in the CV, obtained from a fecal pool of broiler chicken collected from a commercial chicken farm in Pará, Amazon Region, Northern Brazil.
MATERIALS AND METHODS
COLLECTION OF FECAL SPECIMENS
The specimens were collected from a commercial farm located in the Municipality of Benevides, in the metropolitan area of Belém City, capital of Pará State, in Northern Brazil, in July 2009. The pools of fecal samples deposited on beds were collected from nine distinct points in the shed. Those pools refer to 8,460 clinically healthy chickens of the Gallus gallus species, representing the average of 10.8 chickens per square meter, aged 41 days. After collection, the fecal specimens were kept refrigerated in sterile containers, sent to the laboratory, and stored at -70 ºC until processed.
SAMPLE PREPARATION AND VIRAL NUCLEIC ACID EXTRACTION
The fecal suspension was prepared at 10.0% in 0.01 M Tris-Ca++ pH 7.2 buffer, homogenized, and clarified by centrifugation at 700 × g for 20 min at 4 ºC. To obtain the RNA, the fecal pool was extracted using the method reported by Boom et al.7. The RNA was eluted in ultrapure water (free of DNase and RNase) and stored at -70 ºC until further use.
LIBRARY CONSTRUCTION AND TRUSEQ ILLUMINA SEQUENCING
The metagenome sequencing process started with the second strand of the ssRNA. Synthesis was performed using the cDNA Synthesis System kit (Roche Diagnostics) and 400 μM of random primers (Roche Diagnostics, Mannheim, Germany). The reaction was purified with Agencourt AMPure XP reagent (Beckman Coulter, Pasadena, USA). The genome was recovered using a read synthesis system. The cDNA library was prepared and sequenced using the methodology described in the Nextera XT DNA Library Preparation Kit on a Miniseq (Illumina, Inc., San Diego, USA)8.
DATA ANALYSIS
The reads were trimmed to remove the regions with low quality. For genomic assembly, a hybrid approach was followed using the methodology of de novo assembly and reference mapping. The de novo assembly was analyzed using the IDBA-UD program9, and reference mapping was performed using Geneious v.8.1.9 software10. Kraken software11 was used for comparison with other organisms.
PHYLOGENETIC ANALYSIS
The most appropriate evolutionary model was selected using IQ-TREE v.1.3.212. Phylogenetic reconstruction was performed using the maximum likelihood method. Phylogenetic analysis of individual genes and analysis of whole genomes was performed with FastTree v.2.1 software13. The non-parametric bootstrap test with 1,000 replicates was used as the statistical method of reliability. The phylogenetic tree was constructed using Web Server software EvolView14.
RESULTS
HIGH-THROUGHPUT DNA SEQUENCING
High-throughput DNA sequencing generated 2,761,286 high-quality reads, which, following the de novo assembly methodology, resulted in the assembly of 9,737 extension contigs of 328-50,512 bp. Of these, 15 contigs (69-454 bp) were related to members of the Caliciviridae family. The analysis by reference mapping generated a single unit of 292 reads associated with the chicken calicivirus (ChCV) strain RS/BR/2015 (NC_033081)15, demonstrating a nucleotide identity of 93.4% with this strain. The average coverage was 5x, and the GC content was 54.5%. In the analysis of the whole genome sequences, 24.0% to 93.4% homologies and a high divergence with the best-known genera of CV were observed (Figure 1).
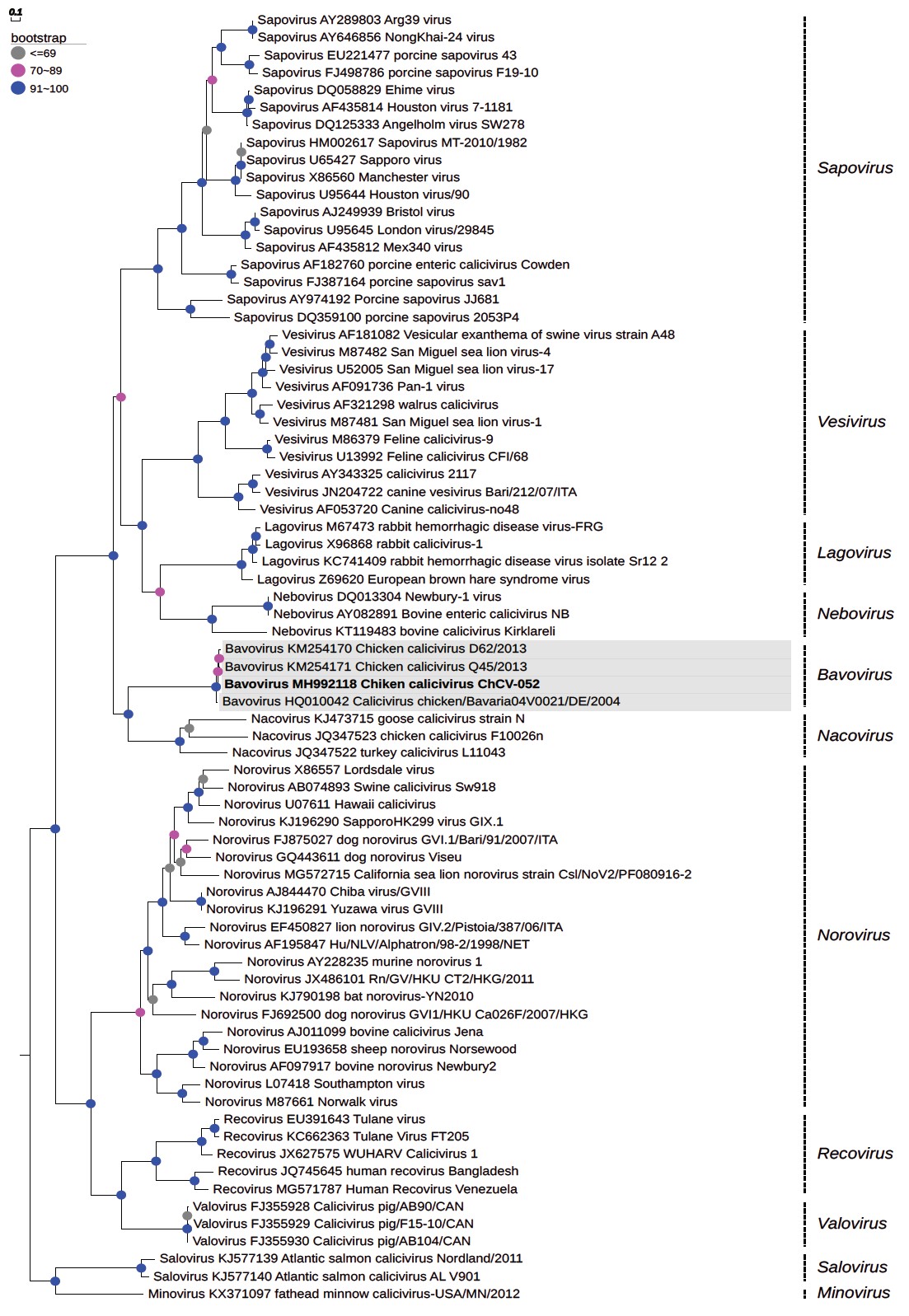
Phylogenetic reconstruction performed by maximum likelihood method, using the non-parametric bootstrap test with 1,000 replicates and constructed by Web Server software EvolView.
Figure 1 - Phylogenetic tree based on the viral capsid region of the genome of Bavovirus (MH992118) detected in a commercial chicken farm from Brazil
CHCV-052 GENOME ANALYSIS
The whole genome size of the ChCV-052 strain was 8.3 kb with a composition of 21.6% A, 23.9% T, 30.0% C, and 24.5% G. The genomic organization of the Caliciviridae family is variable; the genome of strain ChCV-052 exhibited in its extension two ORF: ORF1 containing 6,942 bp (encodes a non-structural polyprotein and major capsid protein - VP1) and ORF2 with 864 bp (encodes a minor capsid protein - VP2).
The characteristic conserved amino acid (AA) motifs of CV were identified as follows: in the RNA-dependent RNA polymerase (RdRp) region, motif DYSGWDST at position 1457, motif GLPSG at position 1512, and motif YGDD at position 1560; in the protease region, motif GDCGLP at position 1176; and in the NTPase region, motif GPPGIGKT at position 345. The GLPSG and YGDD motifs of RdRp were conserved and identical compared to the ChCV RS/BR/2015 strain.
PHYLOGENETIC RELATIONSHIPS
Phylogenetic analysis was based on the whole genome sequences of selected reference strains from the eleven genera of the family Caliciviridae to compose the tree (Figure 2). Phylogeny confirmed that the ChCV-052 strain is grouped with the RS/BR/2015 strain, suggesting its classification into the recently included Bavovirus genus within the Caliciviridae family.
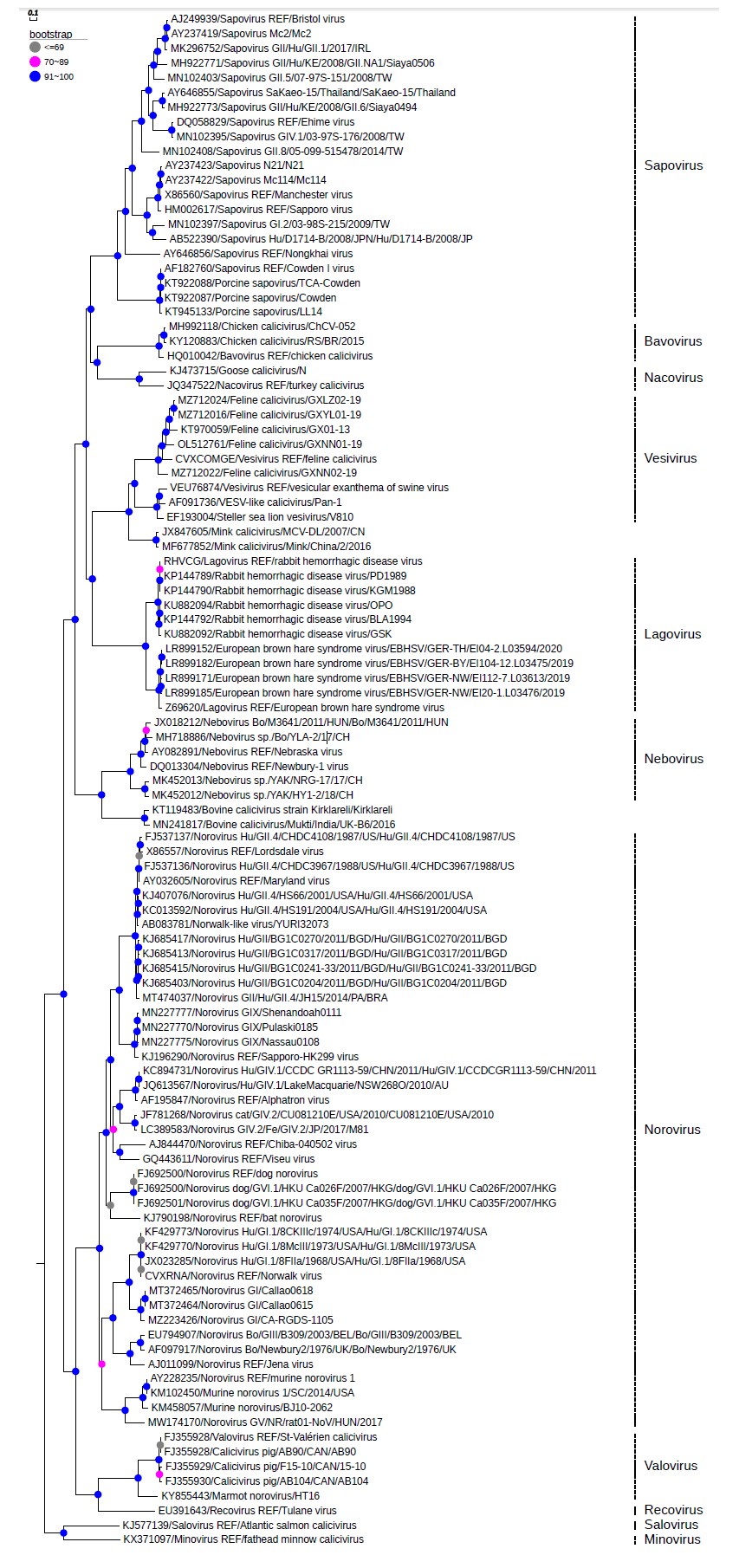
Phylogenetic reconstruction performed by maximum likelihood method, using the non-parametric bootstrap test with 1,000 replicates and constructed by Web Server software EvolView.
Figure 2 - Phylogenetic tree based on the complete genome of Bavovirus (MH992118) detected in a commercial chicken farm from Brazil
The homology percentages within each genus were as follows: 63.6% for genus Vesivirus, 81.6% for genus Lagovirus, 55.3% for Norovirus genus, 76.7% for genus Nebovirus, and 58.7% for genus Sapovirus. The genus Norovirus had a genetic proximity of 53.7% with strain ChCV-052, a low value compared to the referred genus, which had a value of 55.3%.
Another phylogeny based on the sequences of the capsid (Figure 1) revealed values close to those from the previous analysis within each genus as follows: 100.0% for genus Vesivirus, 100.0% for genus Lagovirus, 86.8% for genus Norovirus, 100.0% for genus Nebovirus, and 100.0% for genus Sapovirus. The strain ChCV-052 grouped with the RS/BR/2015 strain with 95.9% nucleotide identity compared to the studied genus and showed a genetic proximity with genus Vesivirus, but the value was slightly lower (48.0%) compared to the homology percentage of 51.9% within the genus.
CODON USAGE
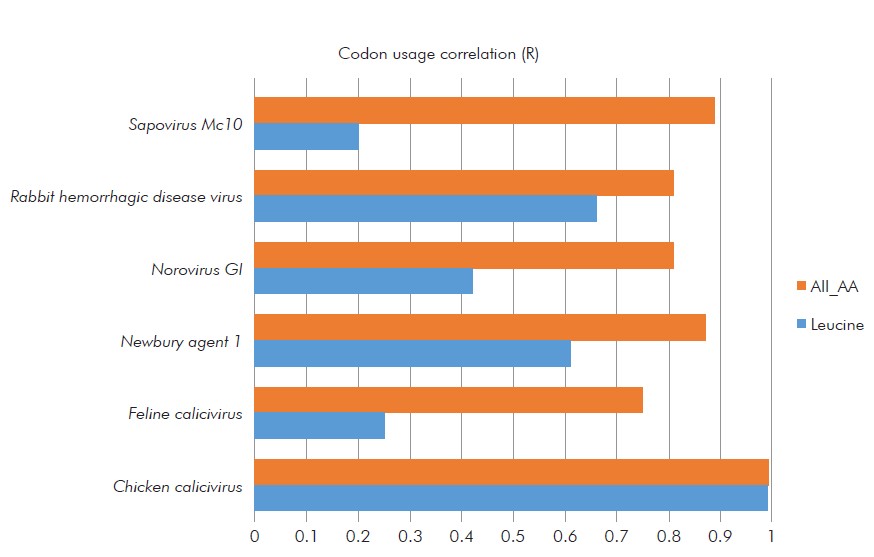
The codon usage bias reflects the balance between mutational pressure and natural selection in the translation optimization process16. There is no data in the literature on codon usage bias in ChCV; therefore, this is the first study that analyzes codon usage in ChCV. The frequency of using synonymous codons for AA coding in the ChCV-052 strain was more correlated with ChCV than with other CV species (Table 1). "Pearson correlation coefficient" showed a higher degree of correlation among ChCV (R= 1) 0.9918 in the coding of all AAs, and 0.99 in the coding of the AA leucine, which composes the genome as one of the most abundant and conserved AAs (Table 2 and Figure 3).
Table 1 - Frequency of use of synonym codons for the AA leucine, encoded in strain ChCV-052 and correlated with ChCV from other calicivirus species
| ChCV-052 | Chicken calicivirus (Unclassified) | Feline calicivirus (Vesivirus) | Newbury agent 1 (Nebovirus) | Norovirus GI (Norovirus) | Rabbit hemorrhagic disease virus (Lagovirus) | Sapovirus Mc10 (Sapovirus) | |
|---|---|---|---|---|---|---|---|
| CTC | 35.50% | 33.30% | 12.40% | 23.30% | 19.80% | 21.80% | 14.40% |
| CTT | 26.90% | 28.00% | 26.60% | 19.50% | 15.50% | 17.50% | 20.50% |
| CTG | 12.00% | 13.60% | 14.70% | 22.80% | 15.90% | 18.90% | 21.90% |
| TTG | 12.00% | 11.50% | 25.70% | 22.80% | 19.30% | 20.90% | 27.90% |
| CTA | 7.40% | 8.20% | 11.90% | 6.50% | 18.40% | 11.20% | 7.90% |
| TTA | 6.20% | 5.30% | 8.70% | 5.10% | 11.10% | 9.70% | 7.40% |
| R | 0.99 | 0.25 | 0.61 | 0.42 | 0.66 | 0.2 |
Table 2 - Codon usage bias showing the balance between mutational pressure and natural selection in the translation optimization process, considering the coding of all AA and the leucine, using the correlation coefficient [Pearson-R], among different caliciviruses
| Chicken calicivirus? (Unclassified) | Feline calicivirus (Vesivirus) | Newbury agent 1 (Nebovirus) | Norovirus GI (Norovirus) | Rabbit hemorrhagic disease virus (Lagovirus) | Sapovirus Mc10 (Sapovirus) | |
|---|---|---|---|---|---|---|
| Leucine | 0.99 | 0.25 | 0.61 | 0.42 | 0.66 | 0.2 |
| All_AA | 0.9918 | 0.75 | 0.87 | 0.81 | 0.81 | 0.89 |
?In this group is inserted the strain ChCV-052, described in the present study.
DISCUSSION
Caliciviruses have been identified in chickens and other birds, such as goldfinch, guinea fowl, pheasant, white tern, and goose17,18. The present study confirms the presence of this virus in chicken flocks, evidencing a genomic signature that is highly distinct compared to the most known CV. Identification of the ChCV-052 strain in chickens corroborates the studies by Wolf et al.19 and Lima et al.15, which detected strains of ChCV from the Bavovirus genus. Sequences of ChCV strains originating in the Netherlands, Germany, Korea, and Brazil deposited in GenBank suggest a widespread distribution of this viral species.
The evolution of genomic composition in CV with GC content ranging from 43.5-58.5% is poorly understood20. The ChCV-052 strain contains 54.5% GC, which is similar to the ChCV RS/BR/2015 (54.2%) strain detected in Brazil, and corroborates previous reports on mammalian and avian caliciviruses15. Different from our results, the bat CV presented a high GC content of 60.2%20. The influence of some factors has been mentioned in the selection of GC content in viral genomes, including host body temperature, immune pressure, and codon and nucleotide usage patterns20.
In addition, the nucleotide composition (GC content) is related to the use of codons20. Our results regarding codon usage showed that the ChCV-052 strain has a coding pattern extremely similar to others ChCV strains, and corroborates the fact that the GC content composition was similar between the ChCV strains (ChCV-052 and RS/BR/2015). This finding reflects a direct correlation between GC content and codon usage. However, there may not be a direct correlation in the increase of GC content with the usage bias codon, as observed in bat CV20. The study by Tse et al.20 suggests that nucleotide composition is the driving force behind codon usage, i.e., it has a direct correlation with codon usage patterns.
The strain in the present study did not had significant mutational changes in its nucleotide composition, which influenced the use of synonymous codons; however, in another study21, the feline CV, ORF2 gene codon use was influenced by mutational bias.
The motif identified in the protease region (present as GDCGLP-ChCV-052) when compared to the motif of ChCV RS/BR/2015 strain (present as GDCGXP) showed an indefinite AA in the sequence; in the NTPase region (present as GPPGIGKT-ChCV-052) compared to the RS/BR/2015 (present as GXPGXGKT), it presented two indefinite AAs in the sequence.
In the RdRp region, the third motif (present as DYSGWDST-ChCV-052) when compared to the RS/BR/2015 strain (present as DYSKWDST) showed a different AA, similar to the one of the other ChCV strain (present as DYSKWDST)17. Therefore, we observed few changes in the AA sequence in CV strains isolated from the same host species (chicken).
This third motif (RdRp) was also identified in baitfish CV (present as DFAAWDKS)22, in goose CV (present as DYKKWD)18, and bat CV (present the DFSKWDST)23 suggesting that CV strains isolated from different host species may exhibit more AA changes that may not alter the functionality of the gene in the infectious process. The other motifs of RdRp (GLPSG and YGDD) were identical and conserved in the above-mentioned CV strains.
However, a comparative study between RNA and DNA virus polymerases24 shows that viral polymerases have a very conserved nature, which reflects on their essential function, and corroborates our result, which identified characteristic motifs in the RdRp region that are conserved (GLPSG and YGDD), and with changes in AAs (DYSGWDST), is also in agreement with Ng et al.25 mentioning RdRp of the CV that causes rabbit hemorrhagic disease virus (RHDV) and showing that the structure and enzymatic mechanisms are common when compared to other RNA viruses.
Our results generate several questions about the relationship between genomic composition versus codon usage and evolutionary virus-host relationships, indicating the need for future studies, since there are no data on this subject that mention ChCV. At the synonymous level, is the accumulation of G and C associated with codon usage rather than translational selection? In evolutionary relations, which occur between CV and their hosts in phylogenetic positioning based on codon usage, will they combine or be opposites?
Identification of the ChCV strain ChCV-052 showed distinct nucleotide characteristics when compared to the main different species of CV, however the phylogenetic analysis strongly support its inclusion in the recently proposed Bavovirus genus, into the Caliciviridae family. The fact that the ChCV-052 strain has been detected in healthy avian hosts requires further studies of the interaction between the host and pathogen biology, which may influence the virus replication process, thus generating necessary molecular changes that regulate the pathogenicity and adaptation of the virus to a new host.
CONCLUSION
This study provided genomic evidence of the presence of Bavovirus in the fecal sample of broilers. Genetically, the ChCV-052 strain is distinct from other CV genera and has a more remarkable similarity to the RS/BR/2015 strain previously described in Brazil. Screening a greater number of samples is necessary to clarify this agent's epidemiology and confirm this virus' occurrence in broilers, determining their genetic diversity. Further research may also help to reveal the transmission routes and elucidate the clinical and economic significance of Bavovirus in this host. To achieve these goals, one can think about establishing sensitive and specific molecular biology techniques to detect this virus in chickens. This report is the first of this genus infecting domestic animals in the Brazilian Amazon.