Wolf-Hirschhorn syndrome (terminal deletion of the short arm of chromosome 4p): Case report1
Síndrome de Wolf-Hirschhorn (deleção do braço curto do cromossomo 4p): Relato de caso
Regina Célia Beltrão DuarteI; Pablo Vaz Gonçalves BorgesII; Rafael da Silva NovaesII; Raimundo Miranda de CarvalhoII; Glória ColonnelliIII; Marcelo Silveira D'OliveiraIV
INeuropediatra, Hospital Universitário João de Barros Barreto, PA. Professora de Neurologia, Faculdade de Medicina da Universidade Federal do Pará e da Universidade Estadual do Pará
IIInterno em Medicina. Universidade Federal do Pará ]]>
IIIMédica Geneticista, Hospital Santa Casa de Misericórdia do Pará. Professora de Genética, Universidade Estadual do Pará
IVGraduando de Medicina.da Universidade Estadual do Pará UEPA
SUMMARY
OBJECTIVE: to report a case of Wolf-Hirschhorn Syndrome or partial deletion of the short arm of one chromosome 4 and present a brief literature review.
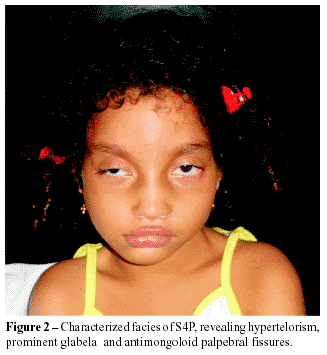
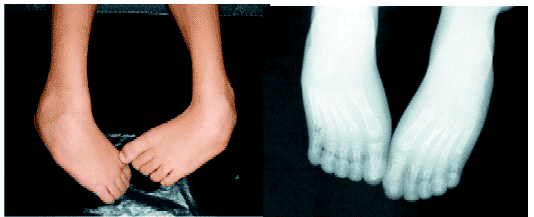
CASE REPORT: the authors report a case of a seven years-old child presenting the main findings of the syndrome: hypertelorism, big and large nose, prominent glabella, high arched eyebrows, antimongoloid palpebral fissures, bilateral low implantation of auricles, and microcephaly. Echodopplercardiographic study evidenced interatrial communication type ostium secundum without hemodynamic repercussion. Radiological examination showed clubfeet. The patient presents many cognitive deficits, mainly in functions as interaction and learning. Evident motor dysfunctions compromising gait and also muscle atrophy. The child also presented convulsive crises in the first year of life that are currently controlled by the use of anticonvulsants.
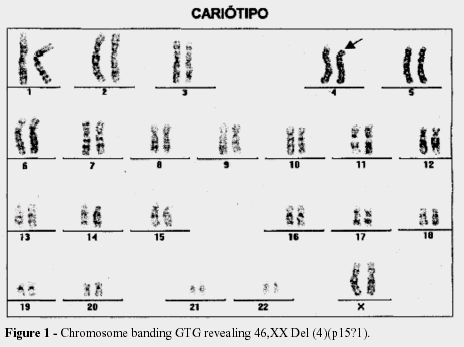
FINAL CONSIDERATIONS: the incidence of the Wolf-Hirschhorn Syndrome is rare, with only 100 cases reported until 1981. The prognosis is relative, with one third of the patients dying with in the first year, while other children are alive with more than twelve years of age. This case was diagnosed based on clinical, radiological, echodopplercardiographical criteria and confirmed by the karyotype's result 46,XX,del(4)(p15?1).
Key-Words: Wolf-Hirschhorn Syndrome, terminal deletion of the short arm of chromosome 4, 4p-, hypertelorism, prominent glabella, IAC.
OBJETIVO: descrever um caso de síndrome de Wolf-Hirschhorn ou deleção parcial terminal do braço curto de um dos cromossomos 4 e apresentar uma breve revisão da literatura.
RELATO DE CASO: criança de sete anos que apresenta os principais aspectos da síndrome: hipertelorismo, nariz grande e adunco, glabela proeminente, fissuras palpebrais antimongolóides, implantação baixa dos pavilhões auriculares, micrognatia e microcefalia. Radiografias do esqueleto revelaram má formação óssea, causando pés eqüinovaros. A ecodopplercardiografia evidenciou CIA sem repercussão hemodinâmica. A paciente apresenta inúmeros déficits cognitivos, fundamentalmente nas funções de interação e aprendizado, além dos distúrbios motores evidentes, com comprometimento da marcha e atrofia muscular; crises convulsivas no primeiro ano de vida, mas que estão atualmente controladas pelo uso de anticonvulsivante.
CONSIDERAÇÕES FINAIS: a incidência da Síndrome de Wolh-Hirschhorn é rara, com 100 casos publicados até 1981. O prognóstico é relativo, com 1/3 dos pacientes morrendo no decorrer do primeiro ano, enquanto outras crianças estão vivas com mais de 12 anos. O caso apresentado foi diagnosticado com base em critérios clínicos, radiológicos, ecodopplercardiogáficos e confirmado pelo resultado do cariótipo: 46,XX,del(4)(p15?1).
Descritores: Síndrome de Wolf-Hirschhorn, deleção do braço curto do cromossomo 4, 4p-, hipertelorismo, glabela proeminente, CIA.
INTRODUCTION
The Wolf-Hirschhorn syndrome (WHS) is a genetic anomaly that is characterized by a group of clinical manifestations, compromising the development and the growth. The disease affects the patient's quality of life and it can be a menace to it, leading to death in the first years. It was described for the first time by Wolff et al. in 1965 and later by Hirschhorn et al. The manifestations are the result of a partial delet ion of the short arm of one chromosome 41.
The incidence is rare (1:50.000), being the female gender more affected (2:1). It has in literature about 100 described cases up to 19811. The clinical features are vast and known: general development deficit, cerebellar and olfactory nerve hypoplasias, agenesis of cerebral structures, hypotonia, seizures, low weight and height (below percentile 3%) and microcefhaly. Facies is characterized by, hypertelorism, big and large nose, micrognathia and others craniofacial abnormalities1,2.
]]> Ocular alterations are frequent such as blepharoptosis, coloboma of iris, divergent strabismus, lachrymal stenosis, antimongoloid palpebral fissures and high arched eyebrows. There are some skeletal features like kyphosis, scoliosis, hemi vertebrae, dysplasia or displacement of the hip, abnormal implantations of hallux and thumb. Also hemangiomas, hypospadia, uterine hipoplasia, hernias, hydronephrosis, polycystic kidney and increased distance between nipples 2,3.Congenital heart defects can be found in WHS such as interatrial communication (IAC) and interventricular communication (IVC). In the literature, there are studies that have found antibodies' deficiency in children affected by WHS, suggesting a tendency to severe infections, especially respiratory ones3,4.
The WHS etiology is known and is related to the deletion of short arm of one chromosome 4, specifically the region 4p16.3. The deletion can occur during the formation of parents' gametes or early in the embryo mitosis and are called de novo. It can be also inherited from parents carrying a balanced translocation. The use of specific techniques (PCR, GTG, fluorescence) may help to identify the correct region deleted of the chromosome 4. The Clinical manifestations of the disease can be noticed since the first years of life, and are important clues to the clinical diagnosis1,3.
Yet there is not a specific treatment for the disease and depending on the clinical features of the patient the treatment can be conducted. However, the life expectation is considered short, especially due to cardio respiratory complications, although there is a case report of a children with 12-years-old4.
OBJECTIVE
To report a case of Wolf-Hirschhorn Syndrome or partial deletion of the short arm of one chromosome 4 and present a brief literature review.
CASE REPORT
Patient L.S.B., female gender, seven years-old, living in Belém-PA. When she was five years old, she was attended at Belém's Pronto-Socorro Municipal presenting vesicles all over her body. Then, the patient was directed to Hospital Universitário João de Barros Barreto in the following day, due to severe clinical condition as dyspnea, jaundice, inferior members paresthesia, fever, cough and nasal congestion.
]]> Previous history of pregnancy, prenatal care and birthHer 28 year-old mother got pregnant, started the prenatal care from the third month (making five consultations), reported vaginal hemorrhage in the fourth month and had received tocolytic agents until the end of the pregnancy. Born of cesarean surgery. At the birth's moment, the patient didn't cry, presented central jaundice, she was taken immediately for the incubator, being at the hospital for eight days with the suspicion of infection. The weight at birth was 2650g.
Personal previous history
At six months of age, she presented jaundice and pneumonia, being hospitalized several times. When she was eight months old, started having no fever seizures, and still has sporadic seizures until now. At nine months, heart murmurs were diagnosed in the left ventricle. Immunization has been brought up to date for her age.
Feeding history
The child was unable to suck and got feed only by baby's bottle, drinking artificial milk until the age of two. At the age of one year-old, she started to drink fruit shake and she started to eat steak six months later. The patient doesn't control the chew. At the breakfast, she used have papaya shake and coffee with milk. At lunch, she eats açai with soup and sticky food. At the afternoon, she has cookies with coffee or shakes and, at night soup.
Growth
The present weight of the patient is 13750g. At nine months of age, there was the eruption of her first tooth and the second dentition has not appeared.
Development
She sustained the head when she was two yearsold, but she doesn't walk and doesn't stand on her feet without support. She whispers to communicate with her parents, saying only the word "water". She presents irregular sleep-wakefulness cycle, sleeping fourteen hours every day.
]]> Familiar previous historyHer 30 year-old father and her 37 year-old mother are healthy. He had already smoked and she had asthma in childhood, but it's in control nowadays. There are arterial hypertension cases in the paternal family. On the mother's side, there are cases of tuberculosis, leprosy, malaria and mental disease.
Physical examination
Good clinical condition, she has no cyanosis or jaundice and her respiratory rate is normal. Microcefhaly, cephalometry of 49 cm and thoracic perimeter of 57 cm; typical face; red-faced skin; normal mucosa; subcutaneous cellular tissue with harmonious distribution, there is no edema and the fold's thickness is normal; muscle hypotrophy of the inferior members; clubfeet; non-palpable ganglia. Orofacial examination: hypertelorism, big and large nose, prominent glabella, antimongoloid palpebral fissures and high arched eyebrows.
Echodopplercardiographic study showed IAC, type ostium secundum of 3,0 mm of extension without hemodynamic repercussion.
The electrocardiogram showed sinus tachycardia. It was done axial computed tomography that showed: hypo density of cerebellar and cerebral's white matter, discrete ectasia of supratentorial ventricular system, especially, occipital portion of lateral ventricles, left putamen infarction and hyper dense area that may correspond to low density calcification of left nucleus's head.
The digital quantitative electroencephalography in induced sleep with chloral hydrate 10% evidenced sequences of slow waves in the right temporal region and left temporal electric activity's depression.
She was seen by a medical geneticist that was responsible for making the karyotype exam that showed the following result 46, XX, Del (4) (p15? 1). Indicating that the child has a partial deletion of a terminal region of the short arm of one chromosome 4. This finding is compatible with the diagnosis of Wolf-Hirschhorn syndrome.
DISCUSSION
]]> This patient's clinical signals are characteristic of WHS, especially those in the face as hypertelorism, big large nose, micrognathia, antimongoloid palpebral fissures, high arched eyebrows and the microcephaly.They are considered as a frequent finding, according to Gonzalez et. al.1.
This child has congenital abnormalities and cytogenetic findings that are described as typical of WHS
Other important signal found in echodopplercardiography study of this child was IAC because is another preponderant abnormality usually found is the presence of cardiovascular alterations, particularly, those located in heart septum, represented by IAC and IVC, according to Bertola et. al.4.
The involvement of neuropsychomotor development is common, including cognitive functions and motor activity5,6,7. In this case report, many cognitive deficits are perceptible, basically functions as interaction and learning. Evident motor dysfunctions with muscle atrophy and compromising gait are present too.
In previous relates, seizures have been described too1. In this study, they occurred when she was one year-old and these crisis are controlled, using anticonvulsants.
According to recent studies8,9,10, 5% to 10% of idiopathic mental retardation's cases are attributed to subtelomeric chromosome deletions, including those which affect the chromosome 4's short arm. This fact happens due to this chromosome region's gene variability and particularitities11,12. The treatment is symptomatic.
High mortality rate (one third of the patients die with in the first year of age), feeding difficulties, muscular hypotonia, seizures (80% of cases), tendency to infections, psychomotor retardation, kyphosis, scoliosis, dentition delay and precocious or delayed puberty The evolution and prognosis of the disease is influenced by the clinical signal.
FINAL CONSIDERATIONS
]]> The diagnostic of genetic disorders can be difficult sometimes because of the heterogeneity of the clinical expression. In this case the diagnostic was important for the parents because they could began the treatment of the symptoms as seizures and had a follow up with physicians and services as neuropediatrics, medical geneticist and others. Besides the parents could have a session of genetic counseling.
REFERÊNCIAS
1. GONZALES, C.H.; WAJNTAL, A.; CAPELOZZI, V.L. Caso em Foco. J. Pediat. São Paulo, 1981, 3: 180-184.
2. HANLEY-LOPEZ, J.; ESTABROOKS, L.L.; STIEHM, R. Antibody deficiency in Wolf-Hirschhorn syndrome. The Journal of Pediatrics. Jul. 1998, 133 (1): 141- 143.
3. BATAGLIA, A.; GALASSO, C.; CEDERHOLM P.; VISKOCHIL D.H.; BROTHMAN, AR. Natural history of Wolf Hirschhorn syndrome: experience with 15 cases. Pediatrics. 1999, 103: 830-836.
4. BERTOLA, D.R.; ALBANO, M.J.; KIM, A.E. Síndromes Genéticas e Cardiopatia. Soc. Cardiol. Estado de São Paulo. 2004, 3: 418-28.
5. SCHLICKUM, S.; MOGHEKAR, A.; SIMPSON, J.C.; STEGLICH, C.; O'BRIAN, R.J.; WINTERPACHT, A. LETM1, a gene deleted in Wolf–Hirschhorn syndrome, encodes an evolutionarily conserved mitochondrial protein. Genomics. 2004, 83: 254–261.
6. BERGEMANN, A.D.; COLE, F.; HIRSCHHORN, K. The etiology of Wolf–Hirschhorn Syndrome. Trends in genetics. March 2005, 21 (3): 188-195.
7. STEVENSON, D.A.; CAREY, C.; BRETT, M.P.H.; COWLEY, B.S.; BROTHMAN, A.R. 4p terminal deletion and 11p subtelomerica duplication detected by genomic microarray in a pacient with Wolf-Hirshhorn Syndrome and an atypical phenotype. J Pediatr. 2004; 145: 840-842.
8. ENDELE, S.; FUHRY, M.; PAK, S.; ZABEL, B.U.; WINTERPACHT, A. LETM1, A Novel Gene Encoding a Putative EFHand Ca21-Binding Protein, Flanks the Wolf–Hirschhorn Syndrome (WHS) Critical Region and Is Deleted in Most WHS Patients. Genomics. 1999, 60: 218–225.
9. WU-CHEN, W.Y.; CHRISTIANSEN, S.P.; BERRY, S.A.; ENGEL, W.K.; FRAY, K.J.; SUMMERS, C.G. Ophthalmic Manifestations of Wolf–Hirschhorn Syndrome. Journal of AAPOS. 4 August 2004, 8 (346): 435-438.
10. WRIGHT, T.J.; COSTA, J.L.; NARANJO, C.; FRANCIS-WEST, P.; ALTHERR, M.R. Comparative Analysis of a Novel Gene from the Wolf–Hirschhorn/Pitt–Rogers–Danks Syndrome Critical Region. Genomics. 1999, 59: 203–212.
11. BORSEL, J.V.; GRANDE, S.D.; BUGGENHOUT, G.V.; FRYNS, J. Speech and language in Wolf-Hirschhorn syndrome: a case-study. Journal of Communication Disorders. 2004, 37: 21–33.
12. TUTUNCULER, F.; ACUNAS, B.; HICDONMEZ, T.; DEVIREN, A.; PELITLI, V. Wolf-Hirschhorn syndrome with posterior intraorbital coloboma cyst: an unusual case. Brain & Development. 2004, 26: 203–205.
 Endereço para Correspondência:
Endereço para Correspondência:
Pablo Vaz Gonçalves Borges.
Rua Oliveira Belo, no 260 - Umarizal
CEP: 66050-380.
Belém – Pará.
Fone: (91) 3241-3779.
E-mail:pablo-vaz@hotmail.com
1- Hospital Universitário João de Barros Barreto/UFPA
]]>